
| 药品信息 | |||
| 受理号: | JXSS1600001 | 药品名称: | 口服五价重配轮状病毒减毒活疫苗(Vero细胞) |
| 药品类型: | 预防用生物制品 | 注册分类: | |
| 承办日期: | 2016-02-22 | 公示日期: | 2021-09-10 |
| 企业名称: | Merck Sharp & Dohme Corp.;Merck Sharp & Dohme Corp.;默沙东研发(中国)有限公司 | ||
| 相关附件信息 | |||
| 附件1:口服五价重配轮状病毒减毒活疫苗(Vero细胞) (JXSS1600001)申请上市技术审评报告.pdf | |||
| 附件2:口服五价重配轮状病毒减毒活疫苗(Vero细胞)(JXSS1600001)-说明书.pdf | |||
核准日期: 修订日期:
口服五价重配轮状病毒减毒活疫苗(Vero细胞)说明书
请仔细阅读说明书并在医师指导下使用
【药品名称】
通用名: 口服五价重配轮状病毒减毒活疫苗(Vero 细胞)
英文商品名: RotaTeq® 中文商品名: 乐儿德 ®
英文名: Reassortant Rotavirus Vaccine, Live, Oral, Pentavalent (Vero Cell)
汉语拼音: Koufu Wujia Chongpei Lunzhuangbingdu Jiandu Huoyimiao (Vero Xibao)
【成份和性状】
淡黄色至略带粉红的淡黄色,澄清液体。
本品为包含 5 种人-牛轮状病毒重配株的口服五价减毒活疫苗, 以 Vero 细胞培养获得。 其中 4 种重配轮状病毒分别表达人轮状病毒亲本株外衣壳蛋白 VP7(血清型 G1,G2 ,G3 或 G4),及牛轮状病毒亲本株黏附蛋白 VP4(血清型 P7[5])。第 5 种重配病毒表达人轮状病毒 亲本株黏附蛋白 VP4(血清型 P1A[8]),及牛轮状病毒亲本株外衣壳蛋白 VP7(血清型 G6) (详见表 1)。
表 1. 重配株的组成
| 重配株 名称 | 人轮状病毒亲本株 和外衣壳蛋白组成 | 牛轮状病毒亲本 株和外衣壳蛋白 组成 | 重配株外衣壳蛋白组成 (人轮状病毒成份用黑体 表示) | 效价不低于 (106 感染单位/剂) |
| G1 | WI79 :G1 ,P1A[8] | WC3 :G6 ,P7[5] | G1 ,P7[5] | 2.2 |
| G2 | SC2 :G2 ,P2A[6] | G2 ,P7[5] | 2.8 | |
| G3 | WI78 :G3 ,P1A[8] | G3 ,P7[5] | 2.2 | |
| G4 | BrB :G4 ,P2A[6] | G4 ,P7[5] | 2.0 | |
| P1A[8] | WI79 :G1 ,P1A[8] | G6 ,P1A[8] | 2.3 |
重配株混悬于稳定缓冲剂中。每一剂疫苗含蔗糖、柠檬酸钠、一水合磷酸二氢钠、氢氧 化钠、聚山梨醇酯 80 ,培养基及痕量的牛血清。不含防腐剂和抗生素。
【接种对象】
6 周至32 周龄婴儿。
【作用与用途】_
本品用于预防血清型 G1 ,G2 ,G3 ,G4 ,G9 导致的婴幼儿轮状病毒胃肠炎。
目前国内注册临床试验尚未观察到足够的 G2 ,G3 ,G4 血清型轮状病毒胃肠炎病例,在 中国健康婴儿中对上述型别的保护作用尚待进一步观察。
【规格】
每支 2ml,每 1 次人用剂量 2ml;所含 5 种人-牛轮状病毒重配株效价不低于如下限值: G1:2.2×106 感染单位 ;G2:2.8×106 感染单位 ;G3:2.2×106 感染单位 ;G4:2.0×106 感染
单位 ;P1A[8] :2.3×106 感染单位。
【免疫程序和剂量】
本品仅限口服,不能用于注射。
本品的全程免疫共3剂:6-12周龄时开始口服第1剂,每剂接种间隔4-10周;第3剂接种 不应晚于32周龄。
【不良反应】
按照国际医学科学组织委员会(CIOMS)推荐的分类描述临床试验中观察到的不良反应的 发生率:十分常见 (≥10%),常见(1%-10%,含 1%),偶见(0.1%-1%,含 0.1%),罕见(0.01%- 0.1%,含 0.01%),十分罕见(<0.01%)。
1. 国外临床试验
本品的 3 项安慰剂对照临床试验对 71,725 名婴儿进行了评估,其中 36,165 名婴儿接种 疫苗,35,560 名婴儿接种安慰剂。
从 11,711 名婴儿(其中 6,138 名接种疫苗)中收集了详细的安全性资料,包括来自注册 试验 006 研究一个亚组和参与 007 等研究的婴儿受试者。在每剂接种后第 1 周内由父母/法 定监护人使用疫苗接种报告卡(VRC)每天记录婴幼儿的体温和任何腹泻及呕吐的发生情况, 并且在每一剂接种后 42 天内用VRC 记录其他不良事件。
在接种本品任何一剂 42 天之内观察到的全身不良反应如下: 十分常见:发热、腹泻、呕吐、易激惹*
常见:胃肠炎* 、肠胃胀气* 、食物反流*、哭闹*
偶见:上呼吸道感染、鼻溢*、便秘*、上腹痛、食欲下降*、咳嗽、鼻咽炎、皮疹、躁 动*、婴儿腹绞痛、失眠*、流感、病毒感染*、鼻塞*、中耳炎*、胃肠道疾病*、腹痛、 便血* 、湿疹* 、激越*、疲劳、肠炎* 、嗜睡* 、尿布性皮炎*、粪便异常、胃部不适、 稀便* 、排便频率增加*、病毒性胃肠炎、呼吸道感染*、乏力*、脱水*
*这些不良反应在疫苗组的发生率低于安慰剂组的发生率。
在本品 III 期临床试验的受试者中,任何一剂接种后 42 天之内未观察到发生频率≥0.1% 的疫苗相关严重不良事件。
肠套叠
本品保护效力和安全性试验(006 研究,也称 REST 试验)研究中,对 34,837 名疫苗受 种者和 34,788 名安慰剂受种者,分别在每剂接种后第 7 、14 和 42 天,以及在第 1 剂接种后 一年内每 6 周对疑似肠套叠病例进行主动监测。
主要安全性评估结果显示,任何一剂接种后 42 天内发生的肠套叠病例,疫苗组有 6 例, 安慰剂组有 5 例(见表 2)。数据表明与安慰剂相比,疫苗未显著增加肠套叠的风险。
表 2. 疫苗组与安慰剂组中肠套叠确诊病例(006 研究)
| 疫苗 (n=34,837) | 安慰剂 (n=34,788) | ||
| 任何一剂接种后42天内出现的肠套叠确诊病例 相对危险度(95% CI)* | 6 | 1.6 (0.4,6.4 ) | 5 |
| 第 1 剂接种后 365 天内出现的肠套叠确诊病例 相对危险度(95% CI) | 13 | 0.9 (0.4,1.9) | 15 |
*在 006 研究中,相对危险度和 95%置信区间以成组序贯设计的终止试验标准为基础。
006 研究中肠套叠病例发生的具体时间见表 3。第 1 剂接种后 42 天内未出现肠套叠确 诊病例,该时间段是人猴重配轮状病毒疫苗(Rotashield® , 已撤市)接种后可能导致肠套 叠发生的最高风险期。
表 3.每剂接种后不同时间段内发生的肠套叠病例(006 研究)
| 第 1 剂 | 第 2 剂 | 第 3 剂 | 任何一剂 | |||||
| 时间范 围 | 疫苗 | 安慰剂 | 疫苗 | 安慰剂 | 疫苗 | 安慰剂 | 疫苗 | 安慰剂 |
| 1-7 天 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| 1-14 天 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 |
| 1-21 天 | 0 | 0 | 3 | 0 | 0 | 1 | 3 | 1 |
| 1-42 天 | 0 | 1 | 4 | 1 | 2 | 3 | 6 | 5 |
在 006 研究中,除一名 9 月龄男孩在第 3 剂接种后第 98 天发生肠套叠后因手术后败血 症死亡外,所有肠套叠患者均痊愈且无后遗症。
在 I 期和 II 期临床研究中,接种本品的 2,470 名受试者中,有 1 名 7 月龄男孩发生肠套 叠,而接种安慰剂的 716 名受试者中未出现肠套叠病例。
川崎病
在 III 期临床试验中,婴儿随访至疫苗接种后 42 天。5/36,150 名疫苗受种者和 1/35,536 名安慰剂受种者报告了川崎病,未校正相对危险度为 4.9(95% CI :0.6 ,239.1)。
便血
在任何一剂接种后 42 天内,0.6%(39/6,130)疫苗受种者和 0.6%(34/5,560)安慰剂受 种者报告了便血的不良事件。在任何一剂接种后 42 天内,<0.1 %(4/36,150)疫苗受种者和 <0.1 %(7/35,536)安慰剂受种者报告了便血的严重不良事件。
惊厥
在本品 III 期临床试验中报告的所有惊厥(按接种组和发生时间)汇总于表 4。
表 4:III 期临床试验任何一剂接种后不同时间段内报告的惊厥
| 时间范围 | 1-7 天 | 1-14 天 | 1-42 天 |
| 疫苗 | 10 | 15 | 33 |
| 安慰剂 | 5 | 8 | 24 |
其中惊厥作为严重不良事件报告的发生率,在疫苗组<0.1%(27/36,150),安慰剂组<0.1% (18/35,536)(无显著差异)。有 10 例高热惊厥报告为严重不良事件,疫苗组 5 例,安慰剂 组 5 例。
2. 国内临床试验
在中国开展的 III 期临床试验共入组 4,040 名 6-12 周龄健康婴儿,按照 1:1 比例随机接 种疫苗或安慰剂。试验中在每剂接种后 30 天内收集体温升高、腹泻和呕吐等征集性不良事 件以及所有其他不良事件(在每一剂接种 14 天内由父母/法定监护人使用VRC 记录婴儿的体 温、任何腹泻和/或呕吐发生情况以及所有其他不良事件,每一剂接种后第 15 天至第 30 天 内出现的所有不良事件由父母/法定监护人通过电话或电子信函收集)。此外, 整个研究期间
收集所有严重不良事件、死亡和肠套叠病例。
在接种任何一剂疫苗后 30 天内,观察到的全身不良反应如下: 十分常见:发热*
常见:腹泻*、呕吐* 、鼻咽炎*
偶见:咳嗽、胃肠炎、咽炎、上呼吸道感染*、肠炎
*这些不良反应在疫苗组的发生率低于安慰剂组的发生率。 接种疫苗的受试者中未出现疫苗相关严重不良事件。
肠套叠
在整个研究期间,报告了2例肠套叠病例,均发生在疫苗组。1例发生在第1剂疫苗接种 后第32天,另1例发生在第3剂疫苗接种后第53天。两例患者经适当治疗均已痊愈。
3. 国外上市后监测
本品上市后在疫苗不良事件报告系统(VAERS)中发现以下不良事件的报告。
向 VAERS 报告接种后发生的不良事件基于自愿,无法估计不良事件的发生频率或确认与 疫苗暴露间的因果关系。
在上市后使用中,接种疫苗后出现以下不良事件的报告: 免疫系统疾病:过敏反应
胃肠道疾病:肠套叠、便血、严重联合免疫缺陷疾病(SCID)患儿发生伴随疫苗株粪便排毒 的胃肠炎
皮肤和皮下组织疾病:荨麻疹、血管性水肿
感染和传染性疾病:川崎病、疫苗株从疫苗受种者传播至未接种疫苗的接触者
【禁忌】
1. 超敏反应:对于本品任何成分出现过敏反应者。
接种一剂本品后出现疑似过敏症状的婴儿,不应继续接种剩余剂次。
2. 严重联合免疫缺陷疾病: 禁止严重联合免疫缺陷疾病(SCID)患儿接种本品。
上市后出现接种疫苗的婴儿发生胃肠炎的报告,包括严重腹泻及疫苗株排毒时 间延长的情况,这些病例随后均被诊断患有SCID。
3. 肠套叠既往史:禁止具有肠套叠既往史婴儿接种本品。
【注意事项】
1. 重要注意事项
(1)过敏反应:必须具备适当的医疗救治和观察条件,以处置接种本品后可能发生的过敏 反应。
(2)免疫缺陷人群
临床试验尚未获得以下潜在免疫缺陷的婴儿服用本品后的安全性和保护效力相关数据:
 患血恶液质、白血病、任何类型淋巴瘤或其他影响骨髓或淋巴系统的恶性肿瘤的婴儿。
患血恶液质、白血病、任何类型淋巴瘤或其他影响骨髓或淋巴系统的恶性肿瘤的婴儿。
 接受免疫抑制剂治疗的婴儿(包括大剂量全身应用皮质类固醇)。对于局部应用皮 质 类固醇或吸入类固醇的婴儿,可以接种本品。
接受免疫抑制剂治疗的婴儿(包括大剂量全身应用皮质类固醇)。对于局部应用皮 质 类固醇或吸入类固醇的婴儿,可以接种本品。
 原发和获得性免疫缺陷婴儿,包括 HIV/AIDS 或人类免疫缺陷病毒感染的其他临床 表现、细胞免疫缺陷、低丙种球蛋白和异常丙种球蛋白血症患者。临床试验中尚无 充分资料支持母亲患有 HIV/AIDS 感染,但 HIV 感染状态尚不确定的婴儿接种本 品。
原发和获得性免疫缺陷婴儿,包括 HIV/AIDS 或人类免疫缺陷病毒感染的其他临床 表现、细胞免疫缺陷、低丙种球蛋白和异常丙种球蛋白血症患者。临床试验中尚无 充分资料支持母亲患有 HIV/AIDS 感染,但 HIV 感染状态尚不确定的婴儿接种本 品。
 42 天内曾输血或血液制品,包括免疫球蛋白的婴儿。
42 天内曾输血或血液制品,包括免疫球蛋白的婴儿。
有报告显示疫苗株可从疫苗受种者传播至未接种疫苗的接触者。(见【注意事项 1.(5)】。
(3)肠套叠
曾在国外上市的人猴重配轮状病毒减毒活疫苗,观察到接种后增加肠套叠风险。
本品在美国上市后观察性研究发现,首剂接种后 21 天内观察到肠套叠病例的发生,病 例集中出现在前 7 天。
本品全球上市后被动监测发现,接种疫苗与肠套叠发生存在时间上的相关性。
(4)胃肠道疾病
对于存在胃肠道功能紊乱史的婴儿,包括活动性急性胃肠道疾病、慢性腹泻和生长发育 迟缓,以及先天性腹部异常和腹部手术史的婴儿,尚无相关的疫苗安全性或保护效力数据。 因此需谨慎考虑对这些婴儿接种本品。
(5)排毒和传播
在本品保护效力和安全性研究(006 研究)中,对其中一个亚组受试者在每一剂接种后 4~6 天,以及在任何时间提供粪便标本的所有受试者中评估了疫苗株粪便排毒的情况。接 种第 1 剂后,32/360 名[8.9%,95% CI(6.2%,12.3%)]疫苗受种者粪便中检测到疫苗株排毒; 接种第 2 剂后,0/249 名[0.0% ,95% CI(0.0% ,1.5%)]疫苗受种者检测到排毒;接种第 3 剂 后,1/385 名[0.3% ,95% CI(<0.1% ,1.4%)]检测到排毒。在 III 期临床研究中,疫苗株排毒 时间最早为接种后第 1 天,最迟为接种后第 15 天,未对疫苗株的传播进行评价。
上市后可观察到本疫苗株从受种者传播至没有接种的接触者。
本疫苗株传播的潜在风险应与感染及传播自然感染轮状病毒的风险进行权衡。 应谨慎考虑是否对与以下免疫缺陷的人密切接触的婴幼儿接种本品:
 恶性疾病或免疫抑制患者;
恶性疾病或免疫抑制患者;
 原发性免疫缺陷患者;
原发性免疫缺陷患者;
 接受免疫抑制剂治疗的患者。
接受免疫抑制剂治疗的患者。
(6)发热性疾病
发热性疾病可能是延迟接种本疫苗的原因,除非医生认为暂停接种本品会造成更大的风 险。
(7)接种不全的免疫程序
临床研究未对仅接种 1 剂或 2 剂本品提供的保护水平进行评估。
(8)疫苗效果局限性:本品可能无法保护所有疫苗受种者感染轮状病毒。
(9)暴露后预防: 尚无关于暴露于自然感染轮状病毒后接种本品的临床数据。
2. 使用方面注意事项:
(1)国外临床研究显示无论在接种本品之前或之后,对于饮食没有限制,包括母乳在内。
(2)如果因某种原因未能给予足够本品剂量(如疫苗被婴儿吐出),由于临床试验中未针对 该情况进行过研究,不建议给予补充接种。应按照后续免疫程序完成剩余剂次的接种。
(3)禁止将本品与其他疫苗或液体混合使用。禁止重配或稀释。
(4)应在有效期内使用本品。
(5)从冰箱中取出本品后,应尽快使用。从冰箱中取出置于 25°C 或低于 25°C 的室温时, 需在 48 小时内使用。超出该期限的产品,应按相关规定弃入生物废弃物容器中。
(6)如果在接种本品前发现其外观与本说明书【成分和性状】描述不一致的变化(如发生 颜色改变等),应避免接种。并按照本说明书提供的联系方式及时进行报告。
(7)本品接种操作说明如下:
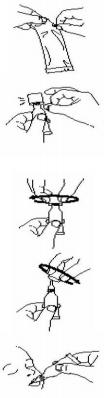
撕开小袋,取出药管。
垂直拿住软管,轻弹管帽,将液体从管口前端弹下。
通过2个简单动作打开定量管:
1. 顺时针旋转管帽直到拧紧,以刺破软管给药口。
2. 逆时针旋转管帽将之取下。
轻轻将液体向婴儿嘴里内颊部挤压,直到挤空软管。(可能在软管顶部 仍有残留液。)
按照相关规定,将空管和管帽弃入生物废弃物容器中。
【儿童用药】
尚未确立本品在小于 6 周龄或大于 32 周龄婴儿中的安全性及保护效力,未批准用于此 类人群。
国外临床研究数据支持胃食管反流病患儿在病情得到良好控制下接种本品。
【药物相互作用】
(1)免疫抑制治疗,包括放射治疗、抗代谢药物、烷化剂、细胞毒性药物及糖皮质激素(大 于生理剂量),可能降低受种者对本品的免疫应答。
(2)疫苗的同时接种
在中国进行的 III 期临床试验数据显示,本品与口服脊髓灰质炎减毒活疫苗(tOPV)和 无细胞百白破疫苗(DTaP)同时接种时,所有不良事件的总体发生率在疫苗组与安慰剂 组间无显著性差异。同时接种本品未显著影响人体对 tOPV 和 DTaP 的免疫原性。
【药物过量】
国外有限的上市后报告本品接种过量(单次接种 1 剂以上疫苗,或食道反流后进行补充 接种)中,由于误用导致过量接种疫苗后观察到的不良事件,与获批的免疫程序和剂量接种 时的情况相似。
【临床试验】
一、国外临床研究
本品国外开展的三个主要III期临床保护效力研究见下表5。
表5. 国外开展的III期临床保护效力研究
| 方案编号 | 样本量 | 受试者年龄范围 | 试验开展的时间地点 |
| 006 [REST] | 69,274 | 6-12 周龄 | 2001 年 1 月-2004 年 10 月;国际多中心(比利 时、哥斯达黎加、芬兰、德国、危地马拉、意 |
| 大利、牙买加、墨西哥、波多黎各、瑞典、台 湾、美国) | |||
| 007 | 1,312 | 6-12 周龄 | 2002 年 9 月-2004 年 6 月;美国、芬兰 |
| 029 | 762 | 6-12 周龄 | 2008 年 8 月-2009 年 8 月;日本 |
以上三个保护效力临床研究中健康婴儿接种3剂,6-12周龄时接种第1剂,后2剂接种间 隔4-10周,32周龄前完成第3剂接种。在这些研究中,不允许同时接种口服脊髓灰质炎疫
苗;但是可以同时接种其他儿童疫苗。所有研究均允许母乳喂养。
在此类保护效力的研究中,轮状病毒胃肠炎病例定义为同时符合以下临床和实验室标 准者:(1)24小时内≥3次水样便或稀便和/或剧烈的呕吐;(2)在如(1)描述的症状出现14 天内采集的粪便中,通过酶免疫分析法(EIA)检测到轮状病毒抗原。
急性轮状病毒胃肠炎的严重程度通过Clark临床评分系统进行评估,评分指标包括发 热、呕吐、腹泻和行为改变的程度及持续时间。
1. 保护效力
临床研究具体结果分别见表 6-8。
表6. 接种后首个轮状病毒流行季内预防任何程度及重度轮状病毒胃肠炎的保护效力
(PPS†)
| 方案编号 | 研究终点 | 严重程度* | 接种人数 | 保护效力,% (95%置信区间) | |
| 疫苗 | 安慰剂 | ||||
| 006 | G1 、G2 、G3 、G4 血清型导致 的轮状病毒胃肠炎 | 任何程度 | 2,834 | 2,839 | 74.0 (66.8, 79.9) |
| 重度 | 2,834 | 2,839 | 98.0 (88.3, 100.0) | ||
| 任何血清型导致的轮状病毒胃 肠炎 | 任何程度 | 2,834 | 2,839 | 71.8 (64.5, 77.8) | |
| 重度 | 2,834 | 2,839 | 98.0 (88.3, 99.9) | ||
| 007 | G1 、G2 、G3 、G4 血清型导致 的轮状病毒胃肠炎 | 任何程度 | 650 | 660 | 72.5 (50.6, 85.6) |
| 重度 | 650 | 660 | 100.0 (13.0, 100.0) | ||
| 任何血清型导致的轮状病毒胃 肠炎 | 任何程度 | 650 | 660 | 72.7 (51.9, 85.4) | |
| 重度 | 650 | 660 | 100.0 (12.7, 100.0) | ||
| 029 | G1 、G2 、G3 、G4 及含 P1A[8]的 G 血清型导致的轮状病毒胃肠 炎 | 任何程度 | 380 | 381 | 74.5 (39.9, 90.6) |
| 重度 | 380 | 381 | 100.0 (55.4, 100.0) | ||
| 任何血清型导致的轮状病毒胃 肠炎 | 任何程度 | 380 | 381 | 75.3 (42.2, 90.9) | |
| 重度 | 380 | 381 | 100.0 (55.2, 100.0) | ||
†PPS(符合方案分析集)包括接种后首个轮状病毒流行季内,第3剂接种至少14天后出现自然感染轮状病毒胃肠炎 病例。
*重度胃肠炎是通过临床评分系统定义,这一评分系统以发热、呕吐、腹泻和行为改变的程度及持续时间为基础。
表7. 接种后首个轮状病毒流行季内预防任何程度及重度轮状病毒胃肠炎的保护效力
(FAS†)
| 方案编号 | 研究终点 | 严重程度* | 接种人数 | 保护效力,% (95%置信区间) | |
| 疫苗 | 安慰剂 | ||||
| 006 | G1 、G2 、G3 、G4 血清型导致的 轮状病毒胃肠炎 | 任何程度 | 2,834 | 2,839 | 60.0 (51.5, 67.1) |
| 重度 | 2,834 | 2,839 | 96.4 (86.2, 99.6) | ||
| 任何血清型导致的轮状病毒胃肠 炎 | 任何程度 | 2,834 | 2,839 | 50.9 (41.6, 58.9) | |
| 重度 | 2,834 | 2,839 | 96.4 (86.3, 99.6) | ||
| 007 | G1 、G2 、G3 、G4 血清型导致的 轮状病毒胃肠炎 | 任何程度 | 650 | 660 | 58.4 (33.8, 74.5) |
| 重度 | 650 | 660 | 100.0 (30.2, 100.0) | ||
| 任何程度 | 650 | 660 | 48.0 (21.6, 66.1) | ||
| 任何血清型导致的轮状病毒胃肠 炎 | 重度 | 650 | 660 | 100.0 (30.4, 100.0) | |
| 029 | G1 、G2 、G3 、G4 及含 P1A[8]的 G 血清型导致的轮状病毒胃肠炎 | 任何程度 | 380 | 381 | 70.3 (32.9, 88.4) |
| 任何血清型导致的轮状病毒胃肠 炎 | 任何程度 | 380 | 381 | 71.3 (35.5, 88.7) |
†FAS(全分析集)包括所有接种至少一剂的受试者中,第1剂接种后首个轮状病毒流行季节内任何时间发生的自然 感染轮状病毒胃肠炎病例。
*重度胃肠炎是通过临床评分系统定义,这一评分系统以发热、呕吐、腹泻和行为改变的程度及持续时间为基础。
表8. 接种后首个轮状病毒流行季预防不同型别导致的任何程度轮状病毒胃肠炎的保护效力
(PPS†)
| 方案编号 | 血清型 | 疫苗 | 安慰剂 | 保护效力,% (95%置信区间) | ||
| 纳入分析人数 | 病例数 | 纳入分析人数 | 病例数 | |||
| 006* | G1P1A[8] | 2,206 | 72 | 2,296 | 286 | 74.9 (67.3, 80.9) |
| G2P1B[4] | 2,204 | 6 | 2,294 | 17 | 63.4 (2.6, 88.2) | |
| G3P1A[8] | 2,203 | 1 | 2,288 | 6 | 82.7 (-42.6, 99.6) | |
| G4P1A[8] | 2,203 | 3 | 2,288 | 6 | 48.1 (-143.2, 91.6) | |
| G9P1A[8] | 2,203 | 1 | 2,287 | 3 | 65.4 (-331.1, 99.3) | |
| ∆ 007 | G1P1A[8] | 551 | 13 | 564 | 53 | 75.8 (55.0, 87.9) |
| G3P1A[8] | 551 | 2 | 562 | 1 | <0 | |
| G9P1A[8] | 551 | 0 | 562 | 1 | 100.0 (-3895.1, 100.0) | |
| 029 | G1P1A[8] | 356 | 3 | 355 | 16 | 81.4 ( 35.1, 96.5) |
| G3P1A[8] | 357 | 4 | 354 | 5 | 20.0 ( -272, 84.1) | |
| G9P1A[8] | 356 | 0 | 354 | 5 | 100.0 ( -9.0, 100.0) | |
| 006、 007 、029 研究合并 分析 | G1P1A[8] | 3,113 | 88 | 3,215 | 355 | 75.3 (68.7,80.7) |
| G2P1B[4] | 3,111 | 6 | 3,210 | 17 | 63.5 (3.0, 88.2) | |
| G3P1A[8] | 3,111 | 7 | 3,204 | 12 | 39.7 (-66.1, 79.9) | |
| G4P1A[8] | 3,110 | 3 | 3,204 | 6 | 48.3 (-142.2, 91.6) | |
| G9P1A[8] | 3,310 | 1 | 3,203 | 9 | 88.5 (17.1, 99.7) | |
| 所有含 P1A[8] 的 G 血清型, 如 G1,G3, G4 和 G9§ | 3,111 | 99 | 3,218 | 383 | 74.2 (67.8, 79.6) | |
注:007 和 029 研究中未观察到 G2 或 G4。
† PPS(符合方案分析集)包括在接种后首个轮状病毒流行季,第 3 剂接种至少 14 天后出现的自然感染轮状病毒胃肠炎 病例。
* 006 研究中,共有 26 例符合方案病例(11 例接种本品,15 例接种安慰剂),其 EIA 结果显示轮状病毒抗原阳性,但是 PCR 结果呈阴性或不可分型。这些病例未被纳入分析。
∆ 007 研究中,有 4 例符合方案病例(1例接种本品,3 例接种安慰剂)的G 血清型未被鉴定(如数据缺失,抗原阳性但是血 清型阴性或不可分型)。这些病例未被纳入分析。
§ 029 研究中,有 1 例病例鉴定为 P1A[8],但是其 G 血清型未被鉴定。该病例纳入此分析。
在006研究中,本品可减少轮状病毒胃肠炎的住院病例,显示了其预防严重疾病的保护 效力。第3剂疫苗接种后两年内,血清型G1 、G2 、G3和G4导致的轮状病毒胃肠炎住院率减 少了95.8%(95% CI :90.5 ,98.2)。减少住院率的FAS保护效力为94.7%(95% CI :89.3,
97.3)。
2. 长期保护效力
006 研究发现疫苗对仅在接种后第二个轮状病毒流行季节出现的 G1、G2、G3 和G4 血清 型导致的任何程度轮状病毒胃肠炎的保护效力为 62.6%(95% CI :44.3 ,75.4),对疫苗接种 后两个轮状病毒流行季中出现的病例的保护效力为 71.3%(95% CI :64.7 ,76.9)。
在芬兰进行的扩展研究(FES)对 006 研究中的 20,736 名受试者开展了接种后 3 年的随 访。006 研究及 FES 研究的汇总分析结果汇总于表 9 。第 3 年,疫苗组未出现轮状病毒胃肠 炎相关就诊病例(n=3,112),安慰剂组出现 1 例(不可分型)(n=3,126)。
表9. 疫苗接种后3年内本品降低轮状病毒胃肠炎导致住院和急诊发生率的保护效力
| 血清型 | G1 | G2 | G3 | G4 | G9 | 任何血清型 |
| 保护效力,% (95%置信区间) | 95.5 (92.8, 97.2) | 81.9 (16.1, 98.0) | 89.0 (53.3, 98.7) | 83.4 (51.2, 95.8) | 94.2 (62.2, 99.9) | 94.0 (91.4, 95.9) |
3. 免疫原性
尚未确定疫苗诱导的抗体应答与预防轮状病毒胃肠炎保护作用之间的相关性。在III期临 床研究中,完成3剂接种后,在疫苗组439名受试者中有92.9%-100%的受试者血清抗-轮状病 毒IgA升高3倍或以上,而在安慰剂组397名受试者中为12.3%-20.0%。
二、国内临床研究
中国的 III 期临床试验为一项随机、双盲、安慰剂对照的多中心研究,旨在评价健康婴儿 接种本品的保护效力、免疫原性与安全性。研究共入组 4,040 名 6-12 周龄健康婴儿,疫苗 组和安慰剂组各 2,020 例。
在此保护效力的研究中,轮状病毒胃肠炎病例定义: (1)受试者在24小时内出现3次或 以上水样便或稀便,和/或剧烈呕吐,(2)在症状出现7天内采集的粪便标本中EIA检出自然感 染野生型轮状病毒。
急性轮状病毒胃肠炎的严重程度通过Vesikari临床评分系统进行评估,评分指标包括呕 吐与腹泻的程度及持续时间,以及发热与脱水的程度和治疗情况。同时采用与国外临床研 究相同的Clark临床评分系统对Clark评分≥17的严重轮状病毒胃肠炎保护效果进行了补充分 析。
1.保护效力
表 10-12 汇总了国内临床研究的保护效力结果。
表 10. 疫苗预防任何程度及重度轮状病毒胃肠炎的保护效力(PPS†)
| 研究终点 | 疫苗 (N = 2,018) | 安慰剂 (N = 2,019) | 保护效力,% (95%置信区间) |
| 病例数 | 病例数 | ||
| 任何血清型导致的任何程度轮状病毒胃肠炎 | 34 | 109 | 69.3 (54.5, 79.7) |
| 任何血清型导致的重度轮状病毒胃肠炎 | |||
| Vesikari评分≥11 | 11 | 52 | 78.9 (59.1, 90.1) |
| Clark评分≥17 | 1 | 22 | 95.5 (71.9, 99.9) |
| G1、G2、G3、G4及含P1A[8]的G血清型引 起的任何程度轮状病毒胃肠炎 | 33 | 108 | 69.9 (55.2, 80.3) |
| G1、G2、G3、G4及含P1A[8]的G血清型引 起的重度轮状病毒胃肠炎 Vesikari评分≥11 Clark评分≥17 | 11 1 | 52 22 | 78.9 (59.1, 90.1) 95.5 (71.9, 99.9) |
N = 接种疫苗或安慰剂的受试者人数。
† PPS(符合方案分析集)包括第 3 剂接种至少 14 天后出现的自然感染轮状病毒胃肠炎病例。
表 11. 疫苗预防任何程度及重度轮状病毒胃肠炎的保护效力(FAS†)
| 研究终点 | 疫苗 (N = 2,018) | 安慰剂 (N = 2,019) | 保护效力,% (95%置信区间) |
| 病例数 | 病例数 | ||
| 任何血清型导致的任何程度轮状病毒胃肠炎 | 35 | 112 | 69.0 (54.4, 79.4) |
| 任何血清型导致的重度轮状病毒胃肠炎 | |||
| Vesikari评分≥11 | 11 | 52 | 79.0 (59.3, 90.1) |
| Clark评分≥17 | 1 | 22 | 95.6 (72.5, 99.9) |
N = 接种疫苗或安慰剂的受试者人数。
† FAS(全分析集)包括所有接种至少一剂的受试者中,第 1 剂接种至少 14 天后出现的自然感染轮状 病毒胃肠炎病例。
表 12. 疫苗预防不同型别导致任何程度轮状病毒胃肠炎的保护效力(PPS†)
| 血清型 | 疫苗 (N = 2,018) | 安慰剂 (N = 2,019) | 保护效力,% (95%置信区间) |
| 病例数 | 病例数 | ||
| G1 | 10 | 39 | 74.4 (47.8, 88.6) |
| G1P1A[8] | 9 | 39 | 77.0 (51.7, 90.2) |
| G1P1B[4] | 1 | 0 | 无法估计 |
| G2 | 1 | 4 | 74.9 (-154.0, 99.5) |
| G2P1B[4] | 1 | 4 | 74.9 (-154.0, 99.5) |
| G3 | 2 | 2 | -0.6 (-1288, 92.7) |
| G3P1A[8] | 2 | 1 | -101.2 (-11772, 89.5) |
| G3P型阴性 | 0 | 1 | 100.0 (-3822, 100.0) |
| G4 | 0 | 2 | 100.0 (-435.4, 100.0) |
| G4P2A[6] | 0 | 2 | 100.0 (-435.4, 100.0) |
| G9 | 20 | 61 | 67.4 (45.2, 81.4) |
| G9P1A[8] | 20 | 61 | 67.4 (45.2, 81.4) |
N = 接种疫苗或安慰剂的受试者人数。
t PPS(符合方案分析集)包括第 3 剂接种至少 14 天后出现的自然感染轮状病毒胃肠炎病例。
国内临床研究结果显示接种本品也可有效预防轮状病毒引起的住院患者。本品预防轮状 病毒胃肠炎住院病例和根据 Vesikari 临床评分系统严重程度≥11 的轮状病毒胃肠炎住院病例 的保护效力分别为 74.0%(95% CI :38.9 ,90.5)和 79.1%(95% CI :44.2 ,93.8)。
2. 免疫原性
尚未确定疫苗诱导的抗体应答与预防轮状病毒胃肠炎保护作用之间的相关性。在中国 III 期临床研究中,接种 3 剂后,疫苗组 349 名受试者中有 89.4%的受试者血清抗-轮状病毒 总 IgA 升高 3 倍或以上,而安慰剂组 356 名受试者中为 10.1%。
3. 安全性
该研究的安全性结果详见【不良反应】部分。
【贮藏】
在 2-8°C 冷藏条件下贮藏及运输。 避光保存。
【包装】
低密度聚乙烯(LDPE)注塑口服软管 1支/盒;10支/盒
【有效期】
24 个月
【执行标准】 【批准文号】
【生产企业】
公司名称:Merck Sharp & Dohme Corp.
地 址:P.O. Box 4, West Point, PA19486, U.S.A
生产厂名称:Merck Sharp & Dohme Corp.
地 址:770, Sumneytown Pike, West Point, PA19486, U.S.A
国内联系方式:
电 话:021 2211 8888
传 真:021 2211 8899
Hits: 618