
受理号:JXSS1800039
药品名称:重组带状疱疹疫苗
药品类型:预防用生物制品
注册分类:
企业名称:GlaxoSmithKline Biologicals SA;GlaxoSmithKline Biologicals SA;葛兰素史克(中国)投资有限公司
承办日期:2018-12-25
公示日期:2020-01-17
核准日期: 修订日期:
重组带状疱疹疫苗(CHO 细胞)说明书
请仔细阅读说明书并在医师指导下使用
【药品名称】
通用名称:重组带状疱疹疫苗(CHO 细胞) 商品名称:欣安立适(SHINGRIX)
英文名称:Recombinant Zoster Vaccine (CHO cell)
汉语拼音:Chongzu Daizhuang Paozhen Yimiao (CHO Xibao)
【成份和性状】
水痘带状疱疹病毒糖蛋白 E(gE)系通过 DNA 重组技术在中国仓鼠卵巢(CHO)细胞中转染截短水痘带状疱疹病毒糖蛋白编码序列,表达特异性抗原, 经纯化、冻干制成。注射用 gE 抗原为无菌白色粉末。
AS01B 佐剂系统混悬液系含有两种免疫增强成分(MPL 和 QS-21)的脂质体制剂。注射用混悬液(AS01B 佐剂系统)为泛乳光的无色到浅褐色液体。 活性成份:
复溶后,1 剂量(0.5 ml)含:
水痘带状疱疹病毒糖蛋白 E (gE) 50μg
佐剂:
AS01B 佐剂系统,1 剂量(0.5ml)含:
皂树皂苷 QS-21 50 μg
3-O-去酰基-4′-单磷酰脂 A(MPL) 50 μg
二油酰基磷脂酰胆碱 1 mg
胆固醇 0.25 mg
其他辅料:
注射用 gE 无菌粉末:蔗糖,聚山梨酯 80,二水合磷酸二氢钠,磷酸氢二钾
注射用混悬液(AS01B 佐剂系统):氯化钠,无水磷酸氢二钠,磷酸二氢钾,注射用水
【接种对象】
本品适用于 50 岁及以上成人。
【作用与用途】
本品适用于预防带状疱疹。 不适用于预防原发性水痘。
【规格】
复溶后每 1 次人用剂量 0.5 ml ,含 gE 蛋白 50 μg。
【免疫程序和剂量】
本品仅限肌肉注射,首选接种部位为上臂三角肌。
免疫程序为两剂,每剂 0.5 ml。第 2 剂与第 1 剂间隔 2 个月接种。
如需改变免疫程序,第 2 剂在第 1 剂后 2~6 个月之间接种(详见【临床试验】)。 尚未确定本品是否需要加强免疫(详见【临床试验】)。
接种前必须复溶疫苗。复溶后,应立即使用疫苗。如果不能立即使用该复溶疫苗应该丢弃。
操作说明:
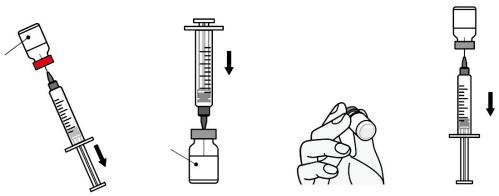
1. 复溶:
| 抗原 注射用无菌粉末 (棕色翻盖小瓶) | 佐剂 注射用混悬液 (蓝绿色翻盖小瓶) |
  用注射器抽取含复溶疫苗 小瓶中的全部内容物。 轻轻振摇, 直至粉末完 全溶解,避免生成气泡。 复溶疫苗为泛乳光、无 色到淡褐色液体。复溶 撕开铝盖,用酒精擦拭 小瓶上部消毒。 使小瓶略微倾斜,将溶 剂小瓶中的全部内容物 抽取至注射器中。 将注射器内的全部内 容物注入含有抗原制 剂的小瓶中。
1 2
| |
| 1 剂量(0.5 ml) |
 1 2
1 2溶剂
1
抗原制剂
 1
1
应按照当地要求立即处置未使用的药品或废料。
2. 接种:
更换针头,使用新针头接种疫苗。
【不良反应】
1.全球临床研究
汇总本品在全球开展的17项临床研究,共有17,041名50岁及以上的成人至少接种了1剂本品。
对本品的安全性评价主要来源于两项安慰剂对照临床研究(ZOSTER-006和ZOSTER-022),这两项研究在北美、拉丁美洲、欧洲、亚洲和澳大利亚 开展,涉及按照0、2月程序接种了至少一剂本品(n=14,645)或生理盐水(n=14,660)的29,305例50岁及以上受试者。受试者首剂接种时的平均年龄为69 岁;7,286例(24.9%)受试者的年龄为50至59岁,4,488例(15.3%)受试者的年龄为60至69岁,17,531例(59.8%)受试者的年龄为70岁及以上。在总人群 中,大多数受试者为白人(74.3%),其次为亚洲人(18.3%)、黑人(1.4%)和其他种族/族裔群体(6.0%);58%为女性。
征集性不良事件
对ZOSTER-006和ZOSTER-022的一个受试者亚组在每次接种疫苗或安慰剂(至少1剂)后7天内使用标准化日记卡收集征集性局部及全身不良反应的 数据,该亚组包括4,886例接种本品及4,881例接种安慰剂的受试者。在这两项研究中,接种本品后报告征集性局部不良反应和征集性全身不良反应的受试 者百分比分别为疼痛(78.0%)、发红(38.1%)和肿胀(25.9%);以及肌痛(44.7%)、疲乏(44.5%)、头痛(37.7%)、寒颤(26.8%)、发热(20.5% )和胃肠道症状(17.3%)。
这两项研究中按年龄组列出的具体征集性局部不良反应和全身不良反应(按受试者统计的总体发生率)的报告率参见表 1 。用于安全性评价的总接 种人群包括具有至少 1 剂接种记录在案的所有受试者(N)。
表1:50至59岁、60至69岁、70岁及以上b成人在接种后7天a 内出现征集性局部不良反应和全身不良反应的受试者百分比(7天日记卡亚组)
| 50 – 59岁 | 60 – 69岁 | ≥ 70岁 | ||||
| 本品 % | 安慰剂c % | 本品 % | 安慰剂c % | 本品 % | 安慰剂c % | |
| 局部不良反应 | N = 1,315 | N = 1,312 | N = 1,311 | N = 1,305 | N = 2,258 | N= 2,263 |
| 疼痛 | 88.4 | 14.4 | 82.8 | 11.1 | 69.2 | 8.8 |
| 疼痛,3级d | 10.3 | 0.5 | 6.9 | 0.5 | 4.0 | 0.2 |
| 发红 | 38.7 | 1.2 | 38.4 | 1.6 | 37.7 | 1.2 |
| 发红,> 100 mm | 2.8 | 0.0 | 2.6 | 0.0 | 3.1 | 0.0 |
| 肿胀 | 30.5 | 0.8 | 26.5 | 1.0 | 23.0 | 1.1 |
| 肿胀,> 100 mm | 1.1 | 0.0 | 0.5 | 0.0 | 1.3 | 0.0 |
| 全身不良反应 | N = 1,315 | N= 1,312 | N= 1,309 | N= 1,305 | N =2,252 | N= 2,264 |
| 肌痛 | 56.9 | 15.2 | 49.0 | 11.2 | 35.1 | 9.9 |
| 肌痛,3级e | 8.9 | 0.9 | 5.3 | 0.8 | 2.8 | 0.4 |
| 疲乏 | 57.0 | 19.8 | 45.7 | 16.8 | 36.6 | 14.4 |
| 疲乏,3级e | 8.5 | 1.8 | 5.0 | 0.8 | 3.5 | 0.8 |
| 头痛 | 50.6 | 21.6 | 39.6 | 15.6 | 29.0 | 11.8 |
| 头痛,3级e | 6.0 | 1.7 | 3.7 | 0.2 | 1.5 | 0.4 |
| 寒颤 | 35.8 | 7.4 | 30.3 | 5.7 | 19.5 | 4.9 |
| 寒颤,3级e | 6.8 | 0.2 | 4.5 | 0.3 | 2.2 | 0.3 |
| 发热 | 27.8 | 3.0 | 23.9 | 3.4 | 14.3 | 2.7 |
| 发热,3级f | 0.4 | 0.2 | 0.5 | 0.2 | 0.1 | 0.1 |
| GI g | 24.3 | 10.7 | 16.7 | 8.7 | 13.5 | 7.6 |
| GI ,3级e | 2.1 | 0.7 | 0.9 | 0.6 | 1.2 | 0.4 |
a 7 天包括接种当天和随后的6 天。
b 50 至 59 岁和 60 至 69 岁的受试者的数据基于 ZOSTER-006 。70 岁及以上受试者的数据基于 ZOSTER-006:NCT01165177 和 ZOSTER-022: NCT01165229 的汇总数据。
c 安慰剂为生理盐水。
d 3 级疼痛:定义为休息时的显著疼痛;阻碍正常的日常活动。 e 3 级肌痛、疲乏、头痛、寒颤、 GI:定义为阻碍正常活动。
f 发热的定义为≥37.5°C/99.5°F(口温、腋温或耳温),或≥38°C/100.4°F(肛温);3 级发热的定义为>39.0°C/102.2°F。 g GI=胃肠道症状,包括恶心、呕吐、腹泻和/或腹痛。
与50至69岁的受试者相比,70岁及以上受试者的征集性局部和全身症状的发生率较低。
接种本品时观察到的大多数征集性局部不良反应和全身不良反应的中位持续时间为2至3天。
在第1剂和第2剂接种后报告的任何或3级征集性局部反应的受试者比例没有差异。第2剂接种后受试者报告头痛和寒颤(分别为28.2%和21.4%),高 于第1剂(分别为24.4%和13.8%)。第2剂接种后受试者报告3级征集性全身不良反应(头痛、寒颤、肌痛和疲乏)(分别为2.3% ,3.1% ,3.6%和3.5%), 高于第1剂(分别为1.4% 、1.4% 、2.3%和2.4%)。
非征集性不良事件
所有受试者在日记卡上记录了每次疫苗接种后30天内(第0天至第29天)发生的非征集性不良事件。这两项研究中接种本品(N=14,645)和安慰剂( N=14,660)的受试者(总接种人群),分别有50.5%和32.0%的受试者报告了疫苗接种后30天内发生的非征集性不良事件。本品受种者中发生率≥1%且比 安慰剂受种者高至少1.5倍的非征集性不良事件包括寒颤(3.5% vs 0.2%)、注射部位瘙痒(2.2% vs 0.2%)、不适(1.7% vs 0.3%)、关节痛(1.7%vs 1.2% )、恶心(1.4%vs 0.5%)和头晕(1.2%vs 0.8%)。
在接种疫苗后的30天内,接种本品和安慰剂的受试者中,痛风(包括痛风性关节炎)的发生率分别为0.18%(N=27)和0.05%(N=8),现有的信息 不足以确定与本品接种存在相关性。
严重不良事件(SAE)
在这两项研究中,从首剂接种至末剂接种后30天,本品和安慰剂受试者的SAE报告率分别为2.3%和2.2%,两组间报告率相似。从首剂接种至末剂接 种后1年,10.1%的本品受试者和10.4%的安慰剂受试者报告了SAE。本品受试者中,1例(<0.01%)报告了淋巴结炎,1例(<0.01%)报告了发热超过39°C ,研究者判断可能与接种本品存在相关性。
潜在的免疫介导疾病(pIMD)
在这两项研究中,从首剂接种至末剂接种后1年,0.6%的本品受试者和0.7%的安慰剂受试者报告了新发的pIMD或现患pIMD恶化。在本品接种组和安 慰剂接种组之间,最常报告的pIMD (≥10例)为风湿性多肌痛、类风湿性关节炎、自身免疫性甲状腺炎和银屑病,两组发生频率相当。
2. 亚洲人群临床研究
在 ZOSTER-006 和 ZOSTER-022 研究中,有 5193 例受试者来自亚洲地区(中国香港和台湾、韩国、日本),并有安全性随访数据。
征集性不良事件
ZOSTER-006 和 ZOSTER-022 的 7 天日记卡亚组中的亚洲人群包括 ZOSTER-006 中的 1,628 例受试者(815 例本品组,813 例安慰剂组)和 ZOSTER-
022 中的 214 受试者(107 例本品组,107 例安慰剂组)。
这两项研究中按年龄组列出的具体征集性局部不良反应和全身不良反应(按受试者统计的总体发生率)的报告频率参见表 2 。用于安全性评价的总 接种人群包括具有至少 1 剂接种记录在案的所有受试者(N)。
表 2:50 岁及以上成人在接种后7 天 a 内出现征集性局部不良反应和全身不良反应的受试者百分比(7 天日记卡亚组的亚洲人群)
| ≥50 岁(ZOSTER-006)b | ≥70 岁(ZOSTER-006 和 ZOSTER-022 的汇总数据)b | |||
| 本品 % | 安慰剂 c % | 本品 % | 安慰剂 c % | |
| 局部不良反应 | N=800 | N=799 | N=400 | N=404 |
| 疼痛 | 83.0 | 14.1 | 74.3 | 13.6 |
| 疼痛,3 级 d | 6.5 | 0.4 | 3.5 | 0.0 |
| 发红 | 45.9 | 2.4 | 45.5 | 2.0 |
| 发红,>100 mm | 4.3 | 0.0 | 5.5 | 0.0 |
| 肿胀 | 37.9 | 1.5 | 38.3 | 1.5 |
| 肿胀,>100 mm | 1.8 | 0.0 | 3.0 | 0.0 |
| 全身不良反应 | N=800 | N=799 | N=400 | N=404 |
| 肌痛 | 57.4 | 13.9 | 41.3 | 13.1 |
| 肌痛,3 级 e | 6.0 | 0.9 | 2.5 | 0.2 |
| 疲乏 | 55.4 | 18.1 | 39.0 | 16.8 |
| 疲乏,3 级 e | 5.9 | 1.0 | 3.5 | 0.7 |
| 头痛 | 40.1 | 12.4 | 27.3 | 12.4 |
| 头痛,3 级 e | 3.4 | 0.3 | 1.5 | 0.5 |
| 寒颤 | 22.4 | 3.6 | 13.5 | 4.5 |
| 寒颤,3 级 e | 2.6 | 0.0 | 1.8 | 0.0 |
| 发热 | 31.6 | 2.6 | 19.5 | 2.7 |
| 发热,3 级 f | 0.3 | 0.1 | 0.3 | 0.0 |
| GIg | 20.4 | 7.6 | 13.8 | 8.2 |
| GI ,3 级 e | 1.0 | 0.1 | 0.8 | 0.2 |
a 7 天包括接种当天和随后的6 天。
b ≥50 岁受试者的数据基于 ZOSTER-006 。70 岁及以上受试者的数据基于 ZOSTER-006(NCT01165177)和 ZOSTER-022(NCT01165229)的汇总数 据。
c 安慰剂为生理盐水。
d 3 级疼痛:定义为休息时的显著疼痛;阻碍正常的日常活动。 e 3 级肌痛、疲乏、头痛、寒颤、GI:定义为阻碍正常活动。
f 发热的定义为≥37.5°C/99.5°F(口温、腋温或耳温),或≥38°C/100.4°F(肛温);3 级发热的定义为>39.0°C/102.2°F。 g GI=胃肠道症状,包括恶心、呕吐、腹泻和/或腹痛。
接种本品时观察到的大多数征集性局部不良反应和全身不良反应的中位持续时间为 1 至 3 天。
非征集性不良事件
在 50 岁及以上亚洲人群受试者中(ZOSTER-006),接种本品 (N=1,432)和安慰剂(N=1,434)的受试者(总接种人群中的亚洲人群)中分别有 52.6% 和 33.8%的受试者报告了疫苗接种后 30 天内发生的非征集性不良事件。≥5%的本品受种者报告的最常见非征集性不良事件为注射部位疼痛(23.5% vs 1.3%)、注射部位肿胀(12.8% vs 0.2%)、发热(11.8%vs 0.8%)和注射部位红斑(9.8% vs 0.1%)。
在70岁及以上受试者中(ZOSTER-006和ZOSTER-022的汇总数据),接种本品(N=1,471)和安慰剂(N=1,472)的受试者(总接种人群中的亚洲亚组) 中分别有56.3%和34.7%的受试者报告了疫苗接种后30天内发生的非征集性不良事件。≥5%的本品受种者报告的最常见非征集性不良事件为注射部位疼痛 (26.0% vs 2.7%)、注射部位肿胀(13.6% vs 0.5%)、注射部位红斑(9.8% vs 0.4%)和发热(7.7% vs 0.7%)。
严重不良事件(SAE)
汇总 ZOSTER-006 和 ZOSTER-022 研究亚洲人群,从首剂接种至末剂接种后 30 天,接种本品和安慰剂的受试者 SAE 报告率分别为 2.4%和 2.7%; 从首剂接种至末剂接种后 1 年,接种本品和安慰剂的受试者 SAE 报告率分别为 10.5%和 10.7%,两组间的报告率相似。
潜在的免疫介导疾病(pIMD)
汇总 ZOSTER-006 和 ZOSTER-022 研究亚洲人群,在整个随访期间,0.7%接种本品的受试者和 0.8%接种安慰剂的受试者报告了pIMD 。
3.全球上市后监测
在本品上市后的使用过程中,已确定了以下不良事件。由于这些事件为规模不详人群的自发报告,因此不一定能够可靠地估计其发生频率或确定其
与疫苗之间的因果关系。 免疫系统疾病
超敏反应,包括血管性水肿、皮疹及荨麻疹。
【禁忌】
对本品的活性成份或任何辅料成份过敏者禁用(详见【成份和性状】)。
【注意事项】
1. 与其他注射用疫苗一样,需准备适当的医疗应急处理措施和监测手段,以保证在接种本品后发生过敏反应者能够及时得到处置。
2. 与其他疫苗一样,在受种者患有急性严重发热疾病时应推迟接种本品。如果仅为感冒等轻微感染,则无需推迟接种。
3. 与其他疫苗相似,接种本品可能无法对所有受种者产生 100%的保护作用。
4. 本品仅用于预防用途,不适用于治疗已发生的临床疾病。
5. 本品严禁静脉或皮内注射。
6. 本品不建议皮下接种。错误地通过皮下接种可能导致一过性局部反应的增加。
7. 本品应慎用于血小板减少症患者或者任何凝血功能紊乱患者,因为这些患者肌肉注射可能发生出血反应。
8. 由于对针剂注射的心因性反应,接种疫苗后或接种前均可能发生晕厥。这种情况可伴随数种神经系统体征,如一过性视觉障碍、感觉异常和强直-阵
挛性肢体活动。为了避免晕厥造成的伤害,应有相应的保护措施。
9. 具有带状疱疹史的个体以及体弱个体(包括患有多种合并症的个体)接种本品的数据有限。因此,医疗专业人员必须基于个体情况权衡接种带状疱
疹疫苗的获益和风险。
10. 免疫抑制人群可能无法产生足够的免疫应答。这些受种者接种本品应谨慎考虑潜在的获益和风险。
11. 接种本品 2-3 天内,可能轻微影响驾驶和操作机器能力。接种后可能会发生疲乏与不适(详见【不良反应】)。
【孕妇及哺乳期妇女用药】 妊娠期用药
目前尚无研究数据评估本品对妊娠期妇女的影响。大鼠、兔生殖毒性研究尚未发现本品对妊娠、胚胎/胎儿发育、分娩或出生后发育造成不良影响。 妊娠期间应避免接种本品。
哺乳期妇女
尚未进行研究评估哺乳期妇女接种本品对母乳喂养婴儿的影响。尚不明确本品是否可经人乳分泌。哺乳期间应避免接种本品。
生育能力
动物研究未发现本品对雄性或雌性动物的生育能力有直接或间接影响。
【药物相互作用】
由于缺少临床研究数据,不建议本品与其他疫苗同时接种。 本品不得与其他药品混合注射。
【临床试验】
(一)全球临床研究结果
1. 对带状疱疹的保护效力
对本品的保护效力评价主要来源于两项安慰剂对照临床研究(ZOSTER-006 和 ZOSTER-022)。ZOSTER-006 研究涉及 15,405 例 50 岁及以上成人, 随机分组间隔 2 个月分别接种两剂本品(7,695 例)或安慰剂(7,710 例); ZOSTER-022 研究涉及 13,900 例 70 岁及以上成人,随机分组间隔 2 个月分别 接种两剂本品(6,950 例)或安慰剂(6,950 例)。
与安慰剂相比,本品对 50 岁及以上成人受试者的带状疱疹保护效力为 97.2% [93.7; 99.0],对 70 岁及以上成人受试者的带状疱疹保护效力为 91.3% [86.8; 94.5],详见表 3。
表 3:本品对带状疱疹的保护效力
| 年龄(岁) | 本品 | 安慰剂 | 保护效力(%) [95 % CI] | ||||
| 可评价受试者人 数 | 带状疱疹病 例数 | 每 1000 人年的发 生率 | 可评价受试者 人数 | 带状疱疹病 例数 | 每 1000 人年的发 生率 | ||
| ZOSTER-006 * | |||||||
| ≥ 50 | 7,344 | 6 | 0.3 | 7,415 | 210 | 9.1 | 97.2 [93.7; 99.0] |
| 50-59 | 3,492 | 3 | 0.3 | 3,525 | 87 | 7.8 | 96.6 [89.6; 99.4] |
| ≥ 60 | 3,852 | 3 | 0.2 | 3,890 | 123 | 10.2 | 97.6 [92.7; 99.6] |
| 60-69 | 2,141 | 2 | 0.3 | 2,166 | 75 | 10.8 | 97.4 [90.1; 99.7] |
| ZOSTER-006 与 ZOSTER-022 汇总 ** | |||||||
| ≥ 70 | 8,250 | 25 | 0.8 | 8,346 | 284 | 9.3 | 91.3 [86.8; 94.5] |
| 70-79 | 6,468 | 19 | 0.8 | 6,554 | 216 | 8.9 | 91.3 [86.0; 94.9] |
| ≥ 80 | 1,782 | 6 | 1.0 | 1,792 | 68 | 11.1 | 91.4 [80.2; 97.0] |
mTVC 排除了未接种第 2 剂或在接种第 2 剂后 1 个月内确诊为带状疱疹的受试者。 CI 置信区间
* 中位随访期 3.1 年 ** 中位随访期 4.0 年
70 岁及以上受试者的数据来自预先定义的 ZOSTER-006 与 ZOSTER-022(mTVC)的汇总分析。
ZOSTER-006 与 ZOSTER-022 研究未排除体弱人群(包括患有多种基础疾病的人群)受试者,该研究包含约 13,000 例患有基础疾病的受试者,包括 带状疱疹高风险相关疾病。对患有常见疾病(冠状动脉疾病、糖尿病、哮喘、 慢性阻塞性肺疾病、抑郁或慢性肾病)并确诊为带状疱疹的患者的保护效 力进行了事后分析,结果表明该效力(84.5%-97%)与总体的预防带状疱疹保护效力相一致。
以上预防带状疱疹的临床研究中提示本品有预防带状疱疹后神经痛的获益,但由于带状疱疹后神经痛例数有限本品在确诊带状疱疹患者中预防 PHN 的效力尚未证实。
2.对带状疱疹的保护效力持久性研究
疫苗接种后第 4 年,对 50 岁及以上成人和 70 岁及以上成人预防带状疱疹的保护效力分别达到 93.1 %(95 %CI:81.2;98.2)和 87.9 %(95 %CI: 73.3 ;95.4)。
目前正在研究 4 年后的保护效力持久性。
3. 免疫程序研究
尚未确定产生保护的免疫学指标,因此预防带状疱疹的免疫应答水平仍不明确。
酶联免疫吸附测定(ELISA)的抗-gE 抗体数据用于支持免疫程序探索。
一项 III 期、开放性临床研究(ZOSTER-026)纳入了 238 例50 岁及以上成人受试者,受试者随机分组,接种 2 剂本品(间隔 2 个月或 6 个月),0 月、 6 月接种程序后的体液免疫应答不劣于 0 月、2 月接种程序后的体液免疫应答。
(二)亚洲人群结果
所有分析都来源于 ZOSTER-006(50 岁及以上受试者 2729 例)及 ZOSTER 006 和 ZOSTER-022 汇总(70 岁及以上受试者 2723 例)的亚洲人群(中 国香港和台湾、日本、韩国)。与安慰剂相比,本品对 50 岁及以上亚洲受试者的带状疱疹保护效力为 95.55 [86.42; 99.11],对 70 岁及以上亚洲受试者的 带状疱疹保护效力为 94.71 [85.89; 98.60],详见表 4。
表 4:本品对带状疱疹的保护效力-亚洲人群亚组
| 年龄 (岁) | 本品 | 安慰剂 | 保护效力(%) [95 % CI] | ||||
| 可评价 受试者 人数 | 带状疱 疹病例 数 | 每 1000 人 年的发生 率 | 可评价受试 者人数 | 带状疱疹 病例数 | 每 1000 人 年的发生 率 | ||
| ZOSTER-006 | |||||||
| ≥ 50 | 1357 | 3 | 0.5 | 1372 | 66 | 12.1 | 95.55 [86.42; 99.11] |
| 50-59 | 667 | 1 | 0.4 | 678 | 31 | 11.4 | 96.84 [81.02; 99.92] |
| ≥ 60 | 690 | 2 | 0.7 | 694 | 35 | 12.9 | 94.42 [78.26; 99.35] |
| 60-69 | 409 | 1 | 0.6 | 409 | 23 | 13.8 | 95.74 [73.77; 99.90] |
| ZOSTER006 与 ZOSTER-022 汇总 | |||||||
| ≥ 70 | 1347 | 4 | 0.8 | 1376 | 75 | 14.7 | 94.71 [85.89; 98.60] |
| 70-79 | 1038 | 3 | 0.7 | 1056 | 59 | 14.8 | 94.95 [84.52; 98.99] |
| ≥ 80 | 309 | 1 | 0.9 | 320 | 16 | 14.3 | 93.82 [60.22; 99.85] |
CI 置信区间
70 岁及以上受试者的数据来自预定的 ZOSTER-006 与 ZOSTER-022(mTVC)的汇总分析。
(三)本品在国内尚未获得临床保护效力研究结果。
【贮藏】
于 2℃-8℃避光保存和运输。 不得冻结。
复溶后: 复溶后疫苗应立即使用。
【包装】
每个包装盒中包括 1 瓶单剂量 gE 无菌粉末和 1 瓶单剂量 AS01B 注射用混悬液。
每个包装盒中包括 10 瓶单剂量 gE 无菌粉末和 10 瓶单剂量 AS01B 注射用混悬液。
【有效期】
36 个月。
【执行标准】
进口药品注册标准:
【进口药品注册证号】:
【生产企业】
企业名称:GlaxoSmithKline Biologicals SA
生产地址:Parc delaNoire Epine, 20 Avenue Fleming, 1300 Wavre, Belgium,比利时 电 话:+32-2-656 81 11
传 真:+32-2-656 80 00
驻中国办事处:上海市西藏中路 168 号都市总部大楼 6 楼; 邮编:200001
电 话:86 21-23019800 传 真:86 21-23019801
GSK 服务热线:800-820-3383/400-183-3383 商标为葛兰素史克集团拥有或授权使用。
© [2018]葛兰素史克集团或其授权人。
Hits: 523