
Contents
RSV疫苗 AREXVY FDA
AREXVY (Respiratory Syncytial Virus Vaccine, Adjuvanted) suspension for intramuscular injection
Initial U.S. Approval: 2023
处方信息的重点
这些重点不包括使用所需的所有信息
安全有效。请参阅完整的处方信息AREXVY。
肌肉注射用AREXVY(含佐剂的呼吸道合胞病毒疫苗)混悬液
美国首次批准时间:2023年
—————适应症和用法——————
AREXVY是一种疫苗,用于主动免疫预防60岁及以上老年人呼吸道合胞病毒引起的下呼吸道疾病(LRTD)。(1)
——————剂量和用法—————
仅用于肌肉注射。
肌肉注射单剂量(0.5毫升)。(2.1)
——————剂型和强度—————
注射用混悬剂以冻干抗原成分的单剂量小瓶形式提供,与随附的佐剂混悬剂成分小瓶一起复溶。复溶后的单次剂量为0.5毫升。(3)
—————禁忌症—————
对疫苗的任何成分有严重过敏反应(如过敏反应)史。(4)
—————不良反应—————
•最常报告的请求局部不良反应(≥10%)是注射部位疼痛(60.9%)。(6.1)
•最常报告的征集的全身不良反应(≥10%)是疲劳(33.6%)、肌痛(28.9%)、头痛(27.2%)和关节痛(18.1%)。(6.1)
要报告疑似不良反应,请联系 葛兰素史克公司1-888-825-5249或VAERS公司1-800-822-7967或www.vaers.hhs.gov。
有关患者咨询信息,请参见第17节。
修订日期:2023年5月
| 完整的处方信息:内容* | 8.2哺乳 *未列出完整处方信息中省略的章节或小节。 |
完整的处方信息
1适应症和用法
AREXVY适用于主动免疫预防60岁及以上老年人呼吸道合胞病毒引起的下呼吸道疾病(LRTD)。
2剂量和用法
2.1剂量和时间表
肌肉注射单剂量(0.5毫升)AREXVY。
2.2准备工作
AREXVY分两瓶提供,接种前必须混合。通过将冻干的抗原成分(无菌白色粉末)与随附的佐剂混悬剂成分(乳白色、无色至淡褐色无菌液体)复溶来制备AREXVY。仅使用随附的佐剂混悬剂成分进行复溶。重组疫苗应为乳白色、无色至淡褐色液体。注射用药品应在注射前目视检查是否有颗粒物质和变色
在溶液和容器允许的情况下给药。如果存在上述任何一种情况,就不应该接种疫苗。
| 图1.清洗两个瓶塞。使用无菌针头和无菌注射器,通过轻微倾斜小瓶,将装有佐剂混悬剂组分(液体)的小瓶中的全部内容物抽出。2瓶中的第1瓶。 | 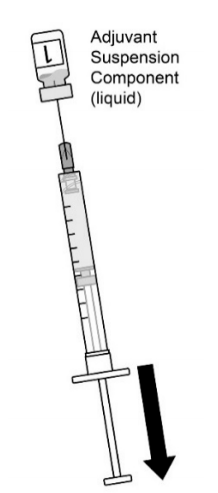 |
| 图2.将注射器的全部内容物缓慢转移到冻干抗原成分小瓶(粉末)中。2瓶中的第2瓶。。 | 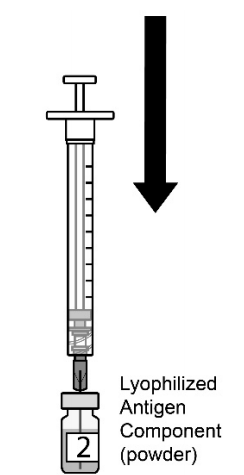 |
| 图3.轻轻旋转小瓶,直到粉末完全溶解。 不要剧烈摇晃。 |  |
| 图4.复溶后,从装有复溶疫苗的小瓶中抽取0.5毫升并进行肌肉注射。 |  |
2.3接种
仅用于肌肉注射。
复溶后,立即接种AREXVY或在2℃至8℃(36℉至46℉)的冰箱中避光保存或在室温下【最高25℃(77℉)】保存,并在4小时内使用。如果未在4小时内使用,请丢弃重组疫苗。
3剂型和强度
AREXVY是一种注射用混悬剂,以单剂量冻干抗原成分小瓶形式提供,与随附的佐剂混悬剂成分小瓶一起复溶。复溶后的单次剂量为0.5毫升。
4禁忌症
请勿将AREXVY用于对AREXVY的任何成分有严重过敏反应(例如过敏反应)史的任何人【参见说明(11)】.
5警告和注意事项
5.1预防和管理疫苗过敏反应
接种AREXVY后,必须进行适当的医疗处理和监督,以控制可能出现的过敏反应。
5.2晕厥
晕厥(昏厥)可能与注射疫苗(包括AREXVY)有关。程序应到位,以避免因昏厥而受伤。
5.3免疫活性改变
免疫功能低下的人,包括接受免疫抑制治疗的人,可能对AREXVY的免疫反应减弱。
6不良反应
在一项临床试验(NCT04886596)中,最常报告的(≥10%)不良反应是注射部位疼痛(60.9%)、疲劳(33.6%)、肌痛(28.9%)、头痛(27.2%)和关节痛(18.1%)。
6.1临床试验经验
由于临床试验是在差异很大的条件下进行的,因此在一种疫苗的临床试验中观察到的不良反应率不能直接与另一种疫苗的临床试验中的不良反应率进行比较,并且可能不能反映实际观察到的不良反应率。
AREXVY的安全性在15,845名疫苗接受者中进行了评估。
研究1(NCT 04886596)是一项安慰剂对照的3期临床研究,在欧洲、北美、亚洲和南半球(南非、澳大利亚和新西兰)进行。涉及24966名60岁及以上的参与者,他们接受了AREXVY(n = 12467)或生理盐水安慰剂(n = 12,499)。研究2(NCT 04732871)是一项在欧洲、北美和亚洲进行的非安慰剂对照、开放标签、3期临床研究,涉及1,653名60岁及以上接受AREXVY的参与者。研究3(NCT 04841577)是一项在新西兰、巴拿马和南非进行的非安慰剂对照、开放标签的3期临床研究,涉及60岁及以上的参与者,他们同时(n = 442)或依次(n = 443)接受1剂AREXVY和FLUARIX四价药。
在研究1中接种疫苗时,人群的中位年龄为69.0岁;13,943名(55.8%)参与者年龄在60至69岁之间,8,978名(36.0%)参与者年龄在70至79岁之间,2,045名(8.2%)参与者年龄在80岁及以上。大多数参与者是白人(79.4%),其次是黑人(8.7%)、亚裔(7.6%)和其他种族/族裔群体(4.3%);5.5%是西班牙裔或拉丁裔;51.7%为女性。在研究2中,接种疫苗时人群的中位年龄为69.0岁;820名(49.6%)参与者年龄在60至69岁之间,621名(37.6%)参与者年龄在70至79岁之间,212名(12.8%)参与者年龄在80岁及以上。在研究2中,大多数参与者是白人(67.8%),其次是亚裔(30.0%)、黑人(2.0%)和其他种族/族裔群体(0.2%);1.9%是西班牙裔或拉丁裔;54.6%为女性。在研究3中,接种疫苗时人口的中位年龄为67.0岁;分别有519名(58.6%)参与者年龄在60至69岁之间,288名(32.5%)参与者年龄在70至79岁之间,78名(8.8%)参与者年龄在80岁及以上。在研究3中,大多数参与者是混血儿(50.3%),其次是白人(30.7%)和黑人(16.0%);34.7%是西班牙裔或拉丁裔;51.5%为女性。
研究1的安全性数据
征集的不良反应:在研究1中,在接种一剂AREXVY或安慰剂后的4天(即接种疫苗当天和接下来的3天)内,使用标准化纸质日记卡监测研究参与者子集(征集安全性集)的征集不良反应;879名参与者接受了AREXVY,874名参与者接受了安慰剂。其他研究参与者没有在日记卡上预先记录征求的反应,但可能将其报告为未经请求的不良反应。
表1列出了特定的局部(给药部位)和全身不良反应(每个参与者)的报告频率。
表1.60岁及以上成人在接种疫苗后4天内出现请求的局部不良反应和全身不良反应的参与者百分比(使用4天日记卡设置的请求安全性)
| 局部不良反应 | AREXVY % N = 879 | 安慰剂a % N = 874 |
| 疼痛,任何b | 60.9 | 9.3 |
| 疼痛,3级b | 1 | 0 |
| 红斑,>20毫米 | 7.5 | 0.8 |
| 红斑,>100毫米 | 0.2 | 0 |
| 膨胀,>20毫米 | 5.5 | 0.6 |
| 膨胀,>100毫米 | 0.2 | 0 |
| 全身不良反应 | N = 879 | N = 878 |
| 疲劳,任何c | 33.6 | 16.1 |
| 疲劳,3级c | 1.7 | 0.5 |
| 肌痛,任何c | 28.9 | 8.2 |
| 肌痛,3级c | 1.4 | 0.3 |
| 头疼,任何c | 27.2 | 12.6 |
| 头痛,3级c | 1.3 | 0 |
| 任何关节痛c | 18.1 | 6.4 |
| 关节痛,3级c | 1.3 | 0.6 |
| 发烧,≥38.0℃/100.4℉ | 2.0 | 0.3 |
| 发烧,>39.0℃/102.2℉ | 0.1 | 0.1 |
N =征求的安全性集合的暴露集合包括至少具有1个记录剂量的所有参与者。
a安慰剂是一种盐溶液。
b .任何级别的疼痛:定义为既不干扰也不妨碍正常日常活动的任何疼痛(1级),当肢体移动并干扰日常活动时疼痛(2级),或休息时明显疼痛并妨碍正常日常活动(3级)。
c .任何级别的疲劳、肌痛、头痛、关节痛:定义为容易耐受(1级)、干扰正常活动(2级)或阻止正常活动(3级)的事件。
通过任何途径(口腔、腋窝或鼓膜)测量体温。
在征求的安全性设置中,AREXVY报告的局部给药部位不良反应的中位持续时间为2天,AREXVY报告的全身不良反应的中位持续时间为1至2天。
主动不良事件:在研究1的所有参与者中,在接种疫苗后的30天内(接种当天和接下来的29天),使用纸质日记卡监测主动不良事件。
在请求安全性组的参与者中,(AREXVY,n = 879或安慰剂,n = 878),在接受AREXVY和安慰剂的参与者中,分别有14.9%和14.6%报告了疫苗接种后30天内发生的主动不良事件。
在暴露组中,24,966名60岁及以上的参与者接受了至少1剂AREXVY(n = 12,467)或安慰剂(n = 12,499)。分别有33.0%和17.8%的参与者报告了在接种疫苗后30天内发生的主动不良事件。与接受安慰剂的参与者相比,接受AREXVY的参与者中报告的主动不良事件的频率更高,这主要归因于与反应原性子集参与者中请求的不良反应一致的事件。
在接种疫苗后30天内,10名接受AREXVY的参与者和4名接受安慰剂的参与者报告了房颤(其中AREXVY组的7起事件和安慰剂组的1起事件为严重事件);接种疫苗后1至30天出现症状。目前关于房颤的可用信息不足以确定与疫苗的因果关系。对于特定类别的主动不良事件,各组之间没有其他值得注意的模式或数字失衡。
严重不良事件:在研究1中,监测参与者在接种AREXVY(n = 12,467)或安慰剂(n = 12,499)后6个月内发生的所有严重不良事件(SAE)。
在接受AREXVY(4.2%)或安慰剂(4.0%)的参与者中,疫苗接种后6个月内发生的SAE的报告率相似。在疫苗接种后6个月内,13名接受AREXVY的参与者和15名接受安慰剂的参与者报告了严重的房颤事件。
死亡人数:从疫苗接种到正在进行的研究1的首次分析,49名接受AREXVY(n = 12,467)的参与者(0.4%)和58名接受安慰剂(n = 12,499)的参与者(0.5%)报告了导致死亡的不良事件。根据现有信息,没有证据表明与甲型肝炎存在因果关系。参与者的死亡原因与成人和老年人群中普遍报告的死亡原因一致。
潜在的免疫介导疾病:在研究1中,对参与者在接种AREXVY(n = 12,467)或安慰剂(n = 12,499)后6个月内发生的所有潜在免疫介导疾病(pIMDs)进行了监测。
在接受AREXVY的参与者中有0.3%和接受安慰剂的参与者中有0.3%报告了疫苗接种后6个月内新发pIMDs或现有pIMDs恶化。在报告的个体pIMDs中,研究组之间没有明显的不平衡。
其他研究报告的严重不良事件
研究2:据报道,在日本的一个研究点登记的一名参与者在接种新冠疫苗后9天开始出现格林-巴利综合征。
研究3:在南非的一个研究中心登记的2名参与者中报告了急性播散性脑脊髓炎;症状分别在接种疫苗后7天和22天出现。
一个事件是致命的,另一个是非致命的。这些参与者同时接受了AREXVY和四价FLUARIX。
8在特定人群中使用
8.1怀孕
风险总结
所有怀孕都有出生缺陷、流产或其他不良后果的风险。在美国普通人群中,临床认可的妊娠中主要出生缺陷和流产的估计背景风险分别为2%至4%和15%至20%。
AREXVY未经批准用于<60岁的人。
在一项临床研究中,与接受安慰剂(用生理盐水复原的蔗糖)的孕妇相比,接受含有与AREXVY相同的RSVPreF3抗原的研究性未添加RSV疫苗的孕妇的早产率增加。
数据
在一项以2:1的比例招募孕妇的随机对照临床试验中,3557名孕妇在孕24至34周时接种了含有与AREXVY相同的RSVPreF3抗原的研究性未添加RSV疫苗,1771名孕妇接种了安慰剂(用生理盐水重构的蔗糖)。在疫苗组和安慰剂组中,分别报告了6.81%和4.95%的早产。
8.2哺乳
风险总结
尚不清楚AREXVY是否会在人乳中排泄。AREXVY未经批准用于<60岁的人。尚无人类或动物数据可用于评估AREXVY对母乳喂养婴儿或乳汁生产/排泄的影响。
母乳喂养的发育和健康益处应与母亲对AREXVY的临床需求以及AREXVY或潜在母体疾病对母乳喂养的孩子的任何潜在不良影响一起考虑。对于预防性疫苗来说,潜在的母体状况是对疫苗预防的疾病的易感性。
8.4儿科使用
来自动物模型的证据强烈表明,AREXVY在2岁以下的个体中不安全,因为增加了呼吸道疾病的风险。尚未确定对2岁至17岁个体的安全性和有效性。
8.5老年用药
AREXVY被批准用于60岁及以上的个人。在总数中在研究1(NCT 04886596)中,接受AREXVY或安慰剂的参与者(N = 24,966)中有13943人(55.8%)年龄在60至69岁之间,8978人(36.0%)年龄在70至79岁之间,2045人(8.2%)年龄在80岁及以上。【参见不良反应(6.1)、临床研究(14.1)】。
11描述
AREXVY(呼吸道合胞病毒疫苗,含佐剂)是一种用于肌肉注射的无菌混悬液。该疫苗以冻干重组呼吸道合胞病毒糖蛋白F(融合前构象稳定)小瓶作为抗原成分提供,在使用时必须与随附的AS01E佐剂小瓶一起重构作为佐剂悬浮液成分。
通过在不含抗生素或动物源性蛋白质的培养基中培养基因工程化的中国仓鼠卵巢细胞来表达RSVPreF3抗原。通过几个色谱和过滤步骤纯化RSVPreF3蛋白,与赋形剂一起配制,装入小瓶并冻干。
AS01E佐剂由3-组成O-去酰基-4’-单磷酰基脂质A(MPL)来自明尼苏达沙门氏菌和QS-21,一种从植物提取物中纯化的皂苷皂树莫利纳,结合在一个脂质体配方。脂质体由二油酰磷脂酰胆碱(DOPC)和胆固醇在含有无水磷酸二钠、磷酸二氢钾、氯化钠和注射用水的磷酸盐缓冲盐水溶液中组成。
复溶后,将每0.5毫升剂量配制成含有120微克重组RSVPreF3抗原、25微克MPL和25微克QS-21。每剂还含有14.7毫克海藻糖、4.4毫克氯化钠、0.83毫克磷酸二氢钾、0.26毫克磷酸二钾、0.18毫克聚山梨醇酯80、0.15毫克无水磷酸二钠、0.5毫克DOPC和0.125毫克胆固醇。
AREXVY不含防腐剂。每剂还可能含有生产过程中残留的宿主细胞蛋白质(≤2.0%)和DNA(≤0.80纳克/毫克)。
瓶塞不是由天然橡胶乳胶制成的。
12临床药理学
12.1作用机制
AREXVY诱导针对RSVpreF3的免疫反应,保护免受RSV引起的LRTD。
13非临床毒理学
13.1致癌、诱变、生育能力受损
尚未评估AREXVY的致癌性或致突变性或对生育能力的损害。
14临床研究
14.1 60岁及以上成人的疗效
研究1(NCT 04886596)评估了AREXVY对60岁及以上成人RSV相关LRTD的疗效。该研究是一项正在进行的3期随机、安慰剂对照、观察者盲临床研究,在北半球和南半球的17个国家开展。计划对参与者进行长达36个月的随访。
这项研究排除了免疫功能低下的参与者。如果研究者认为患有糖尿病、高血压或心脏病等既往慢性稳定疾病的参与者在接种疫苗时身体状况稳定,则允许他们参加研究.
用于疗效分析的主要人群(称为改良暴露组,包括60岁及以上接受1剂AREXVY或安慰剂的成年人,且在接种疫苗后第15天之前未报告RSV确诊的急性呼吸道疾病【ARI】)包括24,960名参与者随机接受1剂AREXVY(n = 12,466)或安慰剂(n = 12,494)。在进行主要疗效分析时,对参与者进行了长达10个月(中位数为6.7个月)的RSV相关LRTD的随访。
在对正在进行的研究1进行首次疗效分析时,51.7%为女性;79.4%是白人,8.7%是黑人,7.6%是亚洲人,4.3%是其他种族/族裔群体,包括美洲印第安人或阿拉斯加土著人、夏威夷土著人或其他太平洋岛民;5.5%是西班牙裔或拉丁裔。参与者的中位年龄为69.0岁。
在基线时,39.3%的参与者至少有一种令人感兴趣的合并症;19.7%的参与者患有基础心肺疾病(慢性阻塞性肺病、哮喘、任何慢性呼吸/肺部疾病或慢性心力衰竭),25.8%的参与者患有内分泌和代谢疾病(糖尿病、晚期肝脏或肾脏疾病)。
抗呼吸道合胞病毒相关下呼吸道疾病的功效
主要目的是证明AREXVY在第一个赛季中预防确诊的RSV-A和/或B相关LRTD首次发作的有效性。
在所有ARI发作期间,通过对鼻咽拭子进行定量逆转录聚合酶链反应(qRT-PCR)来确定确诊的RSV病例。急性呼吸道疾病(ARI)的定义是至少24小时内出现至少2种呼吸道症状/体征(鼻塞、喉咙痛、下呼吸道症状/体征,如下所述),或至少1个呼吸系统症状/体征加上1个全身症状/体征(发热或发烧、疲劳、身体疼痛、头痛、食欲下降)持续至少24小时。LRTD的定义基于以下标准:参与者必须经历过至少2次下呼吸道症状/体征,包括至少24小时内至少1次下呼吸道体征,或至少24小时内经历过至少3次下呼吸道症状。下呼吸道症状包括:新的或增加的痰,新的或增加的咳嗽,新的或增加的呼吸困难(呼吸急促)。下呼吸道症状包括:新出现或加重的喘息、噼啪声/干咳、呼吸频率≥20次/分钟、血氧饱和度低或降低(如果基线<95%,则血氧饱和度≤90%)、需要补充氧气。
与安慰剂相比,在60岁及以上的参与者中,AREXVY显著降低了82.6%的RSV相关LRTD的风险(96.95% CI【57.9,94.1】),达到了主要研究目标的预先指定的成功标准(表2)。疗效随访的中位持续时间为6.7个月。
表2显示了按年龄亚组和至少有一种相关共病的参与者进行的疫苗效力分析。
针对与呼吸道合胞病毒A相关的LRTD病例和与呼吸道合胞病毒B相关的LRTD病例的疫苗效力分别为84.6%(95%可信区间【32.1,98.3】)和80.9%(95%可信区间【49.4,94.3】)。
表2.疗效分析:在研究1中a ,按年龄和共病亚组进行的首次呼吸道合胞病毒相关下呼吸道疾病总体研究(修改暴露设置)
| 子群 | AREXVY | 安慰剂 | 功效百分比(CI)b | ||||
| N | n | 发生率/1,000 人/年 | N | n | 发生率/1,000 人/年 | ||
| 全部的(≥60岁) | 12,466 | 7 | 1.0 | 12,494 | 40 | 5.8 | 82.6 (57.9, 94.1) |
| 60至69岁 | 6,963 | 4 | 1.0 | 6,979 | 21 | 5.5 | 81.0 (43.6, 95.3) |
| 70至79岁 | 4,487 | 1 | 0.4 | 4,487 | 16 | 6.5 | 93.8 (60.2, 99.9) |
| 至少患有1种相关共病的参与者 | 4,937 | 1 | 0.4 | 4,861 | 18 | 6.6 | 94.6 (65.9, 99.9) |
疫苗效力的双边精确置信区间是基于按年龄类别和地区调整的泊松模型得出的。
N =每组包括的参与者人数。
n =从疫苗接种后第15天开始首次出现RSV确诊LRTD的参与者人数。
a研究1: NCT04886596。
b CI =置信区间(总体≥60岁为96.95%,所有亚组分析为95%)。
与安慰剂相比,在70岁及以上的参与者中,AREXVY显著降低了84.4%的RSV相关LRTD风险(95% CI【46.9,97.0】)。由于累计病例总数较少(接受AREXVY的参与者中有2例,接受安慰剂的参与者中有3例),因此无法得出80岁及以上参与者亚组的疫苗有效性(接受AREXVY的参与者中有1,016名参与者,接受安慰剂的参与者中有1,028名参与者)。
对严重呼吸道合胞病毒相关下呼吸道疾病的疗效
在研究1中,严重的RSV相关LRTD被定义为RT-PCR证实的RSV相关LRTD并伴有至少2种下呼吸道症状,或RT-PCR证实的RSV相关LRTD发作阻止正常的日常活动。据报告,接受AREXVY治疗的组中有1例严重的RSV相关LRTD,接受安慰剂治疗的组中有17例,其中2例需要支持治疗。比较的
在接种安慰剂的情况下,AREXVY将60岁及以上参与者发生严重RSV相关LRTD的风险显著降低了94.1%(95% CI【62.4,99.9】)。
14.2合并用药
研究3(NCT 04841577)是一项在新西兰、巴拿马和南非开展的开放性3期临床研究,60岁及以上的参与者接受了1剂AREXVY和第0个月的四价FLUARIX(n = 442)或在第0个月使用1剂fluarix四价疫苗,然后在第1个月使用AREXVY疫苗(n =443)。
没有证据表明对同时使用的两种疫苗中所含任何抗原的免疫反应会受到干扰。对照组与“联合给药”组的免疫反应符合非劣效性标准,因为针对流感病毒A/HongKong/H3N2、流感病毒A/Victoria/H1N1、流感病毒B/Phuket/Yamagata和流感病毒B/Washington/Victoria的RSV-A中和抗体和血凝素抑制抗体的组几何平均滴度比的双侧95%置信区间上限低于1.5。
尚无与其他疫苗同时使用的数据。
16如何供应/储存和搬运
AREXVY以2种成分提供:单剂量瓶装冻干抗原成分(粉末)和单剂量瓶装佐剂悬浮液成分(液体)(包装中无注射器或针头)。
表3AREXVY的产品剂型
| 包装 | 卡尔顿NDC数 | 成分 | |
| 佐剂悬浮液组分(液体) | 冻干抗原成分(粉末) | ||
| 10剂的外盒 | 58160-848-11 | 10瓶 NDC 58160-744-03 | 10瓶 NDC 58160-723-03 |
16.1复溶前的储存
佐剂悬浮液成分小瓶:在2℃和8℃(36℉和46℉)之间冷藏储存。储存在原包装中,以保护小瓶免受光照。不要冻僵。如果出现以下情况则丢弃
佐剂混悬剂成分已被冻结。
冻干抗原成分小瓶:在2℃至8℃(36℉至46℉)之间冷藏储存。储存在原包装中,以保护小瓶免受光照。不要冻僵。如果出现以下情况则丢弃
抗原成分已被冻结。
16.2复溶后的储存
立即接种或在使用前储存在2℃至8℃(36℉至46℉)或室温【最高25℃(77℉)】的冰箱中长达4小时。
保护样品瓶免受光照。
如果未在4小时内使用,丢弃重组疫苗。
不要冻结。如果疫苗已被冷冻,请丢弃。
17患者咨询信息
告知疫苗接种者接种AREXVY疫苗的潜在益处和风险。
告知疫苗接种者接种AREXVY后可能出现的不良反应。
提供疫苗信息声明,可在美国疾病控制和预防中心网站(www.cdc.gov/vaccines)上免费获取。
商标归葛兰素史克集团所有或许可给该集团使用。
制造商葛兰素史克生物制品公司比利时Rixensart美国许可证1617和
分发人葛兰素史克公司
北卡罗来纳州达勒姆市,邮编27701
2023 GSK集团公司或其许可方。ARV:1PI
Hits: 222