
受理号:JXSS1800039
药品名称:重组带状疱疹疫苗
药品类型:预防用生物制品
注册分类:
企业名称:GlaxoSmithKline Biologicals SA;GlaxoSmithKline Biologicals SA;葛兰素史克(中国)投资有限公司
承办日期:2018-12-25
公示日期:2020-01-17
重组带状疱疹疫苗(CHO 细胞)
JX SS1800039
申请上市技术审评报告
国家药品监督管理局药品审评中心 2019 年 9 月
目录
批准 日期:2019 年 5 月 21 日 批准文号:S20190026
重组带状疱疹疫苗(CHO 细胞)
(JXSS1800039)
| 名称 | 地址 | |
| 申请人 | GlaxoSmithKline Biologicals SA | Rue de l`Institut 89, B-1330 Rixensart, Belgium |
| 葛兰素史克(中国)投资有限公司 | 北京市朝阳区东四环中路 56 号楼 A 座 901-910 室 | |
| 生产企业 | GlaxoSmithKline Biologicals SA | Parc de la Noire Epine, 20 Avenue Fleming, 1300 Wavre, Belgium |
| 通用名 | 重组带状疱疹疫苗(CHO 细胞) |
| 英文名 | Recombinant Zoster Vaccine (CHO cell) |
| 剂型及规格 | 注射剂,0.5 mL/剂,每 1 次人用剂量 0.5 mL。 活性成份:每 0.5 mL 剂量约含 50 μg 水痘带状疱疹 病毒糖蛋白 E (gE)。 AS01B 佐剂系统:每 0.5mL 剂量约含 50 μg 皂树皂苷 QS-21,50μg 3-O-去酰基-4′-单磷酰脂 A(MPL),1 mg 二油酰基磷脂酰胆碱,0.25 mg 胆固醇。 |
| 适应症/功能主治 | 本品适用于 50 岁及以上成人预防带状疱疹。不适用 于预防原发性水痘。 |
| 用法用量 | 肌肉注射,首选接种部位为上臂三角肌。 免疫程序为两剂,每剂 0.5 mL。第 2 剂与第 1 剂间 |
| 隔 2 个月接种。如需改变免疫程序,第 2 剂在第 1 剂 后 2~6 个月之间接种。 | |
| 受理的注册分类 | 预防用生物制品 |
| 完成的临床试验内容 | 口Ⅰ期口Ⅱ期口Ⅲ期(境外数据) 其他:国际多中心 |
| 临床试验的合规性 | 临床试验批件号:无 伦理审查批件:口有□无(境外) 知情同意书:口有□无(境外) |
| 特殊审批 | □是 口否 |
| 优先审评 | 口是 □否 |
(略)
NDA 药监局受理日期:2018 年 12 月 12 日
药审中心承办日期:2018 年 12 月 25 日
| 序号 | 会议名称 | 会议时间 |
| 1 | 沟通交流会 | 2018 年 10 月 22 日~23 日 |
| 2 | 专家咨询会 | 2019 年 2 月 21 日~22 日 |
| 3 | 专家咨询会 | 2019 年 4 月 12 日 |
2018 年 8 月本品被列入首批 48 个临床急需境外新药征求意见名单(2018 年 11 月正式发布), 10 月 22 日~23 日药审中心与葛兰素史克(GSK)公司召 开主动沟通交流会。 12 月 12 日本品正式提交进口注册申请, 12 月 25 日药审 中心启动各专业审评任务。2019 年 2 月 3 日完成首轮技术审评,2 月 21 日~22 日召开专家咨询会, 3 月 1 日发出补充资料通知。3 月 11 日~ 4 月 10 日之间, 本品滚动提交回复资料。4 月 12 日召开第二次药学专家咨询会,4 月 15 日启 动专家会后药学专业审评。5 月 6 日收到本品中检院复核检定报告、进口注册
标准及中检院对进口注册标准的复核意见。5 月 16 日,中心完成全部技术审 评,结论为批准生产。
本品被列入 2018 年 11 月 1 日发布的临床急需境外新药名单(第一批)。
本品进口注册申请依据境外临床研究数据,未经过总局审核查验中心现 场核查。
2018 年 12 月 12 日申请人将 3 批成品送样至中国食品药品检定研究院(中 检院)进行进口注册质量标准复核。中检院于 4 月 25 日完成注册检验(中检 生检[2019]456 号),检验结论:本品按拟定进口注册标准检验,结果符合规 定。
本品适用于 50 岁及以上成人,用于预防带状疱疹。
带状疱疹( HZ)是由水痘-带状疱疹病毒(VZV)复苏引起的急性感染 性疾病,好发于老年人和免疫低下人群, 常伴剧烈的顽固性带状疱疹后神经 痛(PHN),严重影响患者生活质量。此外, HZ 还可导致病毒播散、中风、 脑炎和视力损伤(包括失明) 等严重并发症。HZ 发病率较高,研究发现 90% 以上的成人均感染过 VZV,其中约 20%的血清学阳性个体在一生中会发生
HZ。HZ 的治疗包括及时应用抗病毒和镇痛类药物,但这些药物只能减少急 性 HZ 患者的不适及疼痛症状,不能明显的缩短 HZ 的病程以及缓解/减少后 遗症的发生。随着我国社会老龄化加剧,HZ 防控形势更加严峻。
国内尚无带状疱疹疫苗上市,有 5 家企业获得带状疱疹疫苗临床批件, 申报品种均为减毒活疫苗。 国外已有默克公司的 ZOSTAVAX 和 GSK 公司的 SHINGRIX(本品) 两种带状疱疹疫苗获准上市使用。ZOSTAVAX 为全球首 个带状疱疹病毒减毒活疫苗, 于 2005 年 12 月获 FDA 批准上市,主要用于 50 岁以上人群 HZ 的预防。SHINGRIX 为重组灭活带状疱疹疫苗,于 2017 年 10 月获 FDA 批准上市;本品审评时已在美国、欧盟、日本和加拿大批准上市。
本品进行了疫苗佐剂机制研究、佐剂筛选试验和佐剂成分中的 MPL、 QS-21 、DQ 等毒理学研究。
佐剂研究:相比单独给予MPL 、QS-21 ,AS01B 可诱导特异性局部先天免 疫应答;小鼠肌肉注射后 AS01B 迅速分布至引流淋巴结,优先分布在淋巴的 子囊和皮质窦中。与单独使用脂质体、脂质体+MPL、脂质体+QS-21 配制的 gE 相比,用 AS01B 配制的 gE 可诱导小鼠产生更高水平的抗体和CD4+ T 细胞 应答。同一位置注射 gE/AS01B 对 gE 诱导的抗原水平和 CD4+ T 细胞应答水平 较不同位置高,AS01B 和 gE 间隔 0 、1h 在同一位置注射诱导小鼠 gE 特异性 抗体水平与 AS01B/gE 相当,AS01B 和 gE 间隔 6h 内在同一位置注射诱导小鼠 CD4+ T 细胞应答水平与 AS01B/gE 相当。
小鼠肌肉注射 gE/AS01B 诱导产生的 gE 特异性抗体高于皮下注射,细胞 免疫未见明显差异。
( 1 )安全药理学:麻醉雄性大鼠单次静脉注射 gE/AS01B 、AS01B 2 剂/kg, 清醒雄性 Beagle 犬单次肌肉注射 AS01B 1 剂/只,均未见与给药相关的心血管 和呼吸系统毒性。
( 2 )重复给药毒性: 兔连续 4 次、间隔 2 周皮下注射或肌肉注射 gE/AS01B 1 剂/0.5mL/只、AS01B 0.5mL/只佐剂对照,gE/AS01B( SC 、IM)给 药组动物可见一过性纤维蛋白原、 CRP 升高,佐剂对照组动物可见中性粒细 胞增加。 gE/AS01B( SC 、IM)雄兔腘窝淋巴结绝对/相对重量增加。给药均 可见局部刺激性,表现为弥散性混合性炎性细胞浸润。其他未见与受试物相 关的全身毒性。本试验 NOAEL 为 1 剂/只。
兔连续 3 次、间隔 2 周肌肉注射 gE/AS01B 1 剂/0.5mL/只,除可见注射部 位轻微刺激性及几个与局部炎症相关的血液学和血液生化参数一过性异常外, 其他未见明显毒性。本试验 NOAEL 为 1 剂/只。
SD 大鼠连续 7 次、间隔 2 周肌肉注射 AS01B 0.2mL/只,新西兰兔连续 5 次、间隔 2 周肌肉注射 AS01B 0.5mL/只,动物可见局部刺激性及局部炎症和 免疫刺激引起的一过性少数血液学、临床生化参数异常和体温升高,未见明 显全身毒性。
(3 )遗传毒性:雄性SD 大鼠D1 、D2 或单次肌肉注射AS01B 0.2mL/只, 未见对大鼠骨髓红细胞生成能力有明显影响。
( 4 )生殖毒性: 雄性大鼠交配前 42 、28 、14 天肌肉注射 gE/AS01B、
AS01B 1/5 剂/只,gE/AS01B 组动物均诱导产生 gE 抗体,gE/AS01B 、AS01B 组 动物注射部位可见可逆性肿胀,未见 gE/AS01B 、AS01B 对雄鼠生育力及早期 胚胎发育有明显毒性,NOAEL 为1/5 剂/只。雌性大鼠交配前28 、14 天,GD3、
8 、11 、15 天及 LD7 肌肉注射 gE/AS01B 、AS01B 2/5 剂/只,未见对胚胎-胎仔
或产前和子代的生存、生长或发育有明显毒性,NOAEL 为2/5 剂/只。
(5)局部刺激性:兔单次肌肉注射 AS01B 、gE/AS01B 1 剂/只,动物注射 部位可见给药相关的轻微广泛性单核细胞浸润。兔单次皮下注射 AS01B 、 gE/AS01B 1 剂/只,结果 gE/AS01B 、AS01B 组动物分别可见刺激性。
综上,安全性支持本品进行临床试验。 有效性可结合临床试验结果进行 评价。临床需注意给药部位的刺激性;请关注接种部位对免疫原性的影响。
(略)
本品在全球共开展 19 项临床保护效力、免疫原性和安全性研究,其中 17 项在≥50 岁的成人中进行,2 项在免疫功能低下(IC)成人中进行。 19 项临床 研究包括:5 项关键Ⅲ期临床试验、6 项免疫剂量与程序探索研究、6 项免疫 持久性研究, 以及在≥18 岁的 IC 成人中完成的第 Ⅰ/Ⅱ阶段研究和免疫原性探 索研究。 此次注册申请人提供了包含亚洲人群的关键保护效力研究 ZOSTER- 006 、022 及 ZOSTER-006 、022 中的亚洲人群亚组分析, 批间一致性研究 ZOSTER-007,以及免疫程序探索研究 ZOSTER-026(详见表 1 )的详细研究 报告,其余临床试验仅提交了报告摘要。
表 1. 重点关注的关键性Ⅲ期临床试验概要
| 研究 | ZOSTER-006 | ZOSTER-022 | ZOSTER-007 | ZOSTER-026 |
| 研究设计 | 随机、观察者设盲、安慰剂对照的 关键效力研究 | 随机、双盲的批间一致 性研究 | 随机、开放的免疫 程序探索研究 | |
| NCT ID | 01165177 | 01165229 | 02075515 | 01751165 |
| 国家或地区 | 18‡ | 18‡ | 美国、加拿大、比利时 | 美国、爱沙尼亚 |
| 入组例数 | 16160 | 14816 | 651 | 354 |
| 受试者年龄 | ≥50岁 50-59 、60-69 、70-79 和≥80岁,按8:5:3:1 | ≥70岁 70-79岁和≥80岁, 按3:1比例 | ≥50岁 | ≥50岁 |
| 的比例 | ||||
| 主要 研究目的 | ≥50岁成人中预防HZ 的保护效力 | ≥70岁成人中预防 HZ的保护效力 | 批间一致性 | 比较3种免疫程序的 体液免疫反应 |
| 对照组 | 生理盐水 | 生理盐水 | 无 | 无 |
| 分组 | 2组,1:1肌内注射接 种本品或安慰剂 | 2组,1:1肌内注射 接种本品或安慰剂 | 3组,1:1:1肌内注射本 品 | 3组,1:1:1肌内注射 本品 |
| 免疫程序 | 0-2月 | 0-2月 | 0-2月 | 0-2月、0-6月或0-12 月 |
| 随访时间 | 中位3.1年 | 中位数3.9年 | 末次接种后12个月 | |
‡澳大利亚,巴西,加拿大,捷克共和国,爱沙尼亚,芬兰,法国,德国,中国香港,意大利,日本, 韩国,西班牙,瑞典,中国台湾,英国,美国,墨西哥;
4.2.1 临床试验方案
ZOSTER-006 是一项随机、观察者盲、安慰剂对照国际多中心Ⅲ期临床 试验, 旨在评估≥50 岁成人采用 0 、2 个月接种程序肌内接种本品的保护效力、 安全性和免疫原性。该研究在 18 个国家或地区实施,计划入组 15980 例≥50 岁的受试者。主要目的为评估本品与安慰剂相比在≥50 岁的成人中预防 HZ 的 效力。主要假设:证实在≥50 岁的受试者总体预防 HZ 的保护效力(VE)双 侧 95%CI 下限高于 25%。次要目的为评估本品在各年龄层受试者中预防 HZ 的 VE;在总体与各年龄层受试者中预防 PHN 的 VE 等。关于本品预防 PHN 的 VE,审评认为应在≥50 岁的确诊 HZ 受试者中,而非受试者总体中进行评 价。
疑似HZ 病例定义为新发单侧皮疹伴有疼痛(广泛定义包括异常性疼痛、 瘙痒或其他感觉),无替代性诊断。从临床诊断为疑似HZ 病例的受试者皮疹 病变处采集 3 份临床标本,采用标准化和经过验证的 PCR 法对每个皮疹病灶 样本进行检测,按既定流程逐级判定。
本研究同时对接种后细胞免疫(CMI)和体液免疫情况进行了检测。
总接种人群(TVC)包括实际接种疫苗的所有受试者(至少一剂)。调整 总接种人群(mTVC)是效力分析的主要人群, 排除了 TVC 人群中未接种第 2 剂疫苗或在第二次接种后 30 天内发生确诊 HZ 的受试者,或不符合方案规
定和/或给予错误的替代疫苗或给予错误的试验用疫苗的受试者。
研究设置了 7 天日记卡子集、免疫原性子集和细胞免疫原性子集,各子 集样本量详见下表。仅在入选 7 天日记卡子集的受试者中收集接种后第 0 天 至第 6 天的局部或全身征集性 AE,记录在安全性日记卡上;所有其他 AE 均 作为 7 天日记卡子集受试者的非征集 AE 进行记录。对于未纳入 7 天日记卡子 集的受试者,所有 AE 均记录为非征集性 AE。
表 2. ZOSTER-006 中的子集及样本量
| 子集名称 | 参与国家或地区 的数量 | 50-59岁 | 60-69岁 | ≥70岁 | ≥80岁 | 合计 | 总合计 | |||||
| 本品 | 安慰剂 | 本品 | 安慰剂 | 本品 | 安慰剂 | 本品 | 安慰剂 | 本品 | 安慰剂 | |||
| 7天日记卡子集 | 18个国家或地区 | 1410 | 1410 | 1410 | 1410 | 1410 | 1410 | 470 | 470 | 4700 | 4700 | 9400 |
| 免疫原性子集 | 3个国家或地区 (含CMI检测) | 26×3 | 26×3 | 26×3 | 26×3 | 26×3 | 26×3 | — | — | 234 | 234 | 468 |
| 15个国家或地区 (不含CMI检测) | 23×15 | 23×15 | 23×15 | 23×15 | 23×15 | 23×15 | — | — | 1035 | 1035 | 2070 | |
ZOSTER-022 是一项随机、观察者盲、 安慰剂对照的国际多中心Ⅲ期临 床试验, 与 ZOSTER-006 在相同的研究中心同期进行, 旨在评估本品在≥70 岁 老年人中的保护效力,其中 70-79 岁和≥80 岁的受试者比例约为 3:1。计划招 募 14512 例受试者,按 1:1 比例随机分配至试验组或安慰剂组,分别接种 2 剂 (0 、2 月)。主要目的:评估与安慰剂相比本品在≥70 岁受试者中预防 HZ 的 VE。主要假设:在≥70 岁受试者中总体 HZ VE 95%CI 的下限> 10%。次要目 的:在≥70 岁受试者中预防总体 PHN 的VE 等。
如果 ZOSTER-006 和 ZOSTER-022 均证实主要研究终点,则将两项研究 合并的 HZ 和 PHN 数据进行汇总分析,将两项研究中≥70 岁受试者总体 PHN 的汇总数据分析作为 PHN 的主要分析。研究假设:≥70 岁受试者中 HZ VE 95% CI 的下限> 10%,PHN VE 95%CI 的下限>0%。
同 ZOSTER-006,本研究也设置了 7 天日记卡子集和免疫原性子集。70- 79 岁、≥80 岁年龄层的试验组、安慰剂组各 252 例受试者,即共计 1008 例被 随机分配至 7 天日记卡子集。汇总 ZOSTER-006 与 ZOSTER-022,50-59 岁、
60-69 岁、70-79 岁、≥80 岁年龄层的试验组与安慰剂组各 1410 、1410 、1662、 722 例受试者,共计 10408 例被随机分配至7 天日记卡子集。共计 920 例受试 者进入免疫原性子集,这些受试者采血用于评估体液免疫应答。
ZOSTER-007 是一项随机、双盲、多中心(比利时、加拿大和美国的 8 个中心) 的Ⅲ期临床试验,旨在评价三批本品在≥50 岁成人中按第 0 、2 月程 序肌内注射时的一致性、免疫原性、安全性和反应原性。招募 651 例受试者 分为 3 组,按年龄(50-59 岁、60-69 岁和≥70 岁)1:1:1 随机分组,每组 217 例;所有受试者第 0 、3 月采血,通过 ELISA 法检测抗 gE 抗体。主要目的: 证明第 2 剂接种后一个月,本品三个批次之间抗 gE 抗体水平一致性。主要假 设:第 2 剂接种后一个月,本品三个批次两两比较,抗 gE 抗体 GMC 比值的 双侧 95% CI 在[0.67,1.5]之内。次要目的:证明第 2 剂接种后一个月,三个 批次之间抗 gE 抗体阳转率方面的一致性;评价第 0 、3 月时的抗 gE 抗体水 平;评价本品末次接种后一个月和整个研究期间的安全性和反应原性。次要 假设:第 2 剂接种后一个月,本品三个批次两两比较,抗 gE 抗体阳转率差值 的双侧 95%CI 在[-10 %;+ 10 %]以内。
ZOSTER-026 是一项随机、开放、多中心(美国、爱沙尼亚)的Ⅲ期临 床试验, 旨在评价≥50 岁成人中按 0-2 月、0-6 月或 0-12 月接种程序肌内注射 本品的免疫原性和安全性。拟招募 354 例受试者按 1:1:1 随机分组,每组 118 例。所有受试者在基线期(第 0 天)和第 2 剂次接种后 1 个月,通过 ELISA 法检测抗 gE 抗体,并进行安全性观察。主要目的:证明 0-6 月组、0-12 月组 第 2 剂接种后 1 个月抗 gE 抗体非劣效于 0-2 月组。主要假设:第 2 剂接种后 1 个月抗 gE GMC 比值(0-2 月组/0-6 月组或 0-12 月组) 的 97.5%CI 上限应小 于 1.5。次要目的:评估所有组别的抗 gE 体液免疫应答;评估本品的安全性 和反应原性。
4.2.2 临床试验结果
( 1 )临床试验完成情况
ZOSTER-006 研究开始于 2010 年 8 月 2 日,共入组 16160 例受试者。最 终 HZ 保护效力分析(第 1 步)截止日期为 2014 年 7 月 1 日,2014 年 12 月 1 日最终 HZ 保护效力分析的数据库冻结。EOS(End of Study)效力分析(第 2 步)的截止日期为 2015 年 4 月 21 日,2015 年 10 月 12 日 EOS 分析数据库冻 结,同时也是安全性评价的截止日期。采用两步法分析:先在最终 HZ 效力分 析(第 1 步)步骤中分析 HZ VE,然后在 EOS 中(第 2 步)分析 PHN VE 。 在最终 HZ 保护效力分析中, 15411 例受试者被纳入 TVC 人群,其中试验组 7698 例,安慰剂组 7713 例。 14759 例受试者被纳入 mTVC 人群,试验组 7344 例,安慰剂组 7415 例。开展汇总分析时,与 ZOSTER-022 合并 70-79 岁和≥80 岁年龄层接种者的数据。
ZOSTER-022 研究开始于 2010 年 8 月 2 日,EOS 效力分析的截止日期为 2015 年 4 月 21 日,EOS 数据库冻结时间为2015 年 10 月 12 日。共入选 14816 例受试者,13900 例受试者被纳入 TVC 人群,其中试验组6950 例,安慰剂组 6950 例。 13163 例受试者被纳入 mTVC 人群,试验组 6541 例,安慰剂组 6622 例。
ZOSTER-007 研究开始于 2014 年 8 月 13 日,完成于 2015 年 4 月 29 日, 2015 年 8 月 13 日数据库锁定。共招募 651 例受试者,622 例被纳入免疫原性 符合方案人群(免疫原性 ATP 人群)。
ZOSTER-026 研究开始于 2013 年 3 月 12 日,完成于 2014 年 5 月 22 日, 2015 年 3 月 23 日数据库锁定。共招募 354 例受试者,343 例受试者被纳入用 于免疫原性分析的 ATP 人群。
亚洲亚组分析:为支持本品在国内注册,申请人对 ZOSTER-006 和 022 研究中 5196 例来自中国(包括中国台湾和中国香港)、韩国和日本的亚洲亚 组人群的安全性和有效性进行了描述性分析。ZOSTER-006 和022 汇总的TVC
Asian 共有 5193 例受试者,其中试验组 2596 例,安慰剂组 2597 例;共有4524 例受试者完成了研究,其中试验组 2252 例和安慰剂组 2272 例。4886 例受试 者被纳入 mTVC Asian,其中试验组 2423 例和安慰剂组 2463 例。
(2)全球人群有效性数据
① 保护效力方面:
ZOSTER-006 研究对 14759 例≥50 岁受试者进行为期超过 3 年的随访( 中 位随访时间为 3.1 年)。最终 HZ 效力分析阶段,mTVC 人群中共有 216 例受 试者确诊为 HZ 病例,试验组6 例,安慰剂组 210 例;无受试者报告一次以上 的 HZ 发作。总体来讲,89.4%的 HZ 病例通过 PCR 法确诊,10.6%通过 HZAC 的裁定确诊。其中,试验组通过 PCR 法确定了4 例(66.7%),通过 HZAC 确 定了2 例(33.3%);安慰剂组通过PCR 法确定了189 例(90.0%),通过HZAC 确定了 21 例( 10.0%)。
在≥50 岁的受试者中,试验组每 1000 人年的总体 HZ 发生率为 0.3,安慰 剂组为 9.1。按照发病密度计算,本品在≥50 岁的受试者中,对 HZ 的总体保 护率为 97.16%(95% CI:93.72%-98.97%)。按 50-59 岁、60-69 岁、≥70 岁分 层分析,本品对HZ 的保护率在各年龄层相当,其中50-59 岁HZ VE 为96.57% (95% CI:89.62%-99.31%),60-69 岁 HZ VE 为 97.36 %(95 % CI:90.14 %- 99.69 %),≥70 岁 HZ VE 为 97.93 %(95 % CI:87.91 %-99.95 %)。
表 3. 研究期间 HZ VE 的分析(mTVC-最终 HZ 效力分析)
| 本品 | 安慰剂 | VE | P值 | |||||||||
| 年龄分层 | N | n | T(年) | n/T (/ 1000) | N | n | T (年) | n/T (/ 1000) | % | 95 % CI LL | 95 % CI UL | |
| 50-59岁 | 3492 | 3 | 11161.3 | 0.3 | 3525 | 87 | 11134.7 | 7.8 | 96.57 | 89.62 | 99.31 | <0.0001 |
| 60-69岁 | 2141 | 2 | 7007.9 | 0.3 | 2166 | 75 | 6952.7 | 10.8 | 97.36 | 90.14 | 99.69 | <0.0001 |
| ≥70岁 | 1711 | 1 | 5127.9 | 0.2 | 1724 | 48 | 5083.0 | 9.4 | 97.93 | 87.91 | 99.95 | <0.0001 |
| ≥60岁 | 3852 | 3 | 12135.7 | 0.2 | 3890 | 123 | 12035.7 | 10.2 | 97.58 | 92.77 | 99.51 | <0.0001 |
| 总体 | 7344 | 6 | 23297.0 | 0.3 | 7415 | 210 | 23170.5 | 9.1 | 97.16 | 93.72 | 98.97 | <0.0001 |
N=每组受试者人数; n=至少有一次确诊 HZ 发作的受试者人数; T(年)=以年为单位表示的随访期总和 (在首次发生确诊的 HZ 发作时进行删失); n/T(每 1000 )=受试者报告至少一例事件的发生率;LL , UL=95%置信下限和置信上限;VE(%)=疫苗保护效力。
根据方案规定,确诊 HZ 时,首次 VZV PCR 高于 0 个拷贝/PCR 但低于临 界水平, 需对皮疹病变样本重复检测 2 次,以获取 3 个结果。如果至少2 个结 果≥临界水平,则判定为VZV 阳性,反之则判定为VZV 阴性。申请人后续补 充提供了确诊 HZ 病例中进行重复 VZV PCR 的结果。截止 EOS 分析阶段,本 研究共 9 例进行重复 VZV PCR,其中 5 例最终确诊为HZ 病例。考虑到 VZV PCR 复测确诊的HZ 病例占所有 HZ 病例(共 263 例)的比例较小( 1.90%), 排除后不会影响 HZ VE 的整体结论。
在 EOS 分析阶段,试验组确诊 HZ 的 9 例受试者中均没有报告 PHN 发作; 安慰剂组 254 例确诊 HZ 的受试者中有 18 例(7.09%)报告至少 1 起 PHN 发 作。 总体 PHN VE 为 100.00%(95% CI:77.11%-100.00%)。在确诊 HZ 病例 中的 PHN VE 为 100.00%(95% CI:-345.68% – 100.00%)。
ZOSTER-022 研究对 13163 例≥70 岁的受试者,进行为期超过 3 年的随访 (中位随访时间为 3.9 年)。mTVC 人群中共计确诊 246 例 HZ 受试者,其中 试验组 23 例,安慰剂组 223 例,没有受试者报告一次以上的 HZ 发作。 总体 来讲,92.3%的 HZ 病例通过 PCR 法确诊,7.7%通过 HZAC 裁定确定。其中, 试验组中 82.6%的 HZ 病例通过 PCR 法确诊, 17.4%通过 HZAC 裁定确定;
安慰剂组中93.3%的 HZ 病例通过 PCR 法确诊,6.7%通过 HZAC 裁定确定。
在≥70 岁的受试者中,试验组 HZ 总发病率为 0.9/1000 人年,安慰剂组为 9.2/1000 人年。按照发病密度计算,本品在≥70 岁的受试者中,对 HZ 的总体 VE 为 89.79%(95%CI:84.29%~93.66%)。按照 70-79 岁、≥80 岁分层分析, 本品对 HZ 的保护率在两个年龄层相当,其中 70-79 岁:90.02 %(95 % CI: 83.54 %~94.32 %),≥80 岁:89.08 %(95 % CI:74.65 %~96.16 %)。
按性别、地区、种族、时间进行敏感性分析,没有观察到 HZ VE 明显的 差异。采用 Poisson 方法按时间(使用 1 年间隔和累计)分析 HZ VE,第 4 年 的 HZ VE 仍达 85.07%(95%CI:64.47~94.83%)。
表 4.研究期间 HZ VE 的分析 (mTVC)
| 年龄分层 | 本品 | 安慰剂 | VE | |||||||||
| N | n | T (年) | n/T (/1000) | N | N | T (年) | n/T (/1000) | (%) | 95 % CI LL | 95 % CI UL | P 值 | |
| 70-79 岁 | 5114 | 17 | 19346.5 | 0.9 | 5189 | 169 | 19247.5 | 8.8 | 90.02 | 83.54 | 94.32 | <0.0001 |
| ≥80 岁 | 1427 | 6 | 5058.5 | 1.2 | 1433 | 54 | 4920.3 | 11.0 | 89.08 | 74.65 | 96.16 | <0.0001 |
| 总体 | 6541 | 23 | 24405.1 | 0.9 | 6622 | 223 | 24167.8 | 9.2 | 89.79 | 84.29 | 93.66 | <0.0001 |
同 ZOSTER-006 研究,本研究共 6 例进行重复 VZV PCR,其中 2 例最终 确诊为 HZ 病例。VZV PCR 复测确诊的 HZ 病例仅占所有 HZ 病例的 0.8% (2/246),排除后不会影响 HZ VE 的整体结论。
在 EOS 分析中,试验组确诊 HZ 的 23 例受试者中 4 例报告 PHN 发作, 安慰剂组确诊 HZ 的 223 例受试者中28 例报告至少 1 起 PHN 发作。 总体 PHN VE 为 85.49%(95%CI:58.52%~96.30%)。在确诊 HZ 病例中的 PHN VE 为 – 35.63%(95%CI:-222.72% ~49.12%)。
ZOSTER-006 及 ZOSTER-022 汇总分析:两项研究汇总分析共入组了 29305 例≥70 岁的受试者,其中试验组 14645 例,安慰剂组 14660 例。共确诊 309 例 HZ 病例,其中试验组 25 例,安慰剂组 284 例。无受试者报告一次以 上的HZ 发作。在≥70 岁的受试者中,试验组的HZ 总发病率为0.8/1000 人年, 安慰剂组为 9.3/1000 人年,总体HZ VE 为91.30%(95 % CI: 86.88 %~94.46 %) 。 70-79 岁、≥80 岁的 HZ VE 分别为 91.27%% (95 % CI:86.04 %~94.85 %) 、 91.37%(95 % CI:80.22 %~96.94 %),两个年龄层 HZ VE 基本一致。
表 5.研究期间 HZ VE 的分析( mTVC ,≥70 岁)
| 本品 | 安慰剂 | VE | |||||||||||
| 研究 | 年龄分层 | N | n | T (年) | n/T (/1000) | N | n | T (年) | n/T (/1000) | (%) | 95 % CI LL | 95 % CI UL | P 值 |
| ZOSTER- 006/022 汇总 | 70-79 岁 | 6468 | 19 | 24410.9 | 0.8 | 6554 | 216 | 24262.8 | 8.9 | 91.27 | 86.04 | 94.85 | <0.0001 |
| ≥ 80 岁 | 1782 | 6 | 6314.6 | 1.0 | 1792 | 68 | 6151.9 | 11.1 | 91.37 | 80.22 | 96.94 | <0.0001 | |
| ≥70 岁 | 8250 | 25 | 30725.5 | 0.8 | 8346 | 284 | 30414.7 | 9.3 | 91.30 | 86.88 | 94.46 | <0.0001 | |
两项研究汇总分析,在≥50 岁的受试者中共 50 例确诊 HZ 的受试者至少 报告了 1 次 PHN。其中,试验组 32 例 HZ 病例中的 4 例( 12.5%)和安慰剂
组 477 例HZ 病例中的46 例(9.6%)报告了 PHN。在确诊HZ 病例中的 PHN VE 为 0.29 % (95 % CI:-161.53 %~65.57 %), 因此在≥50 岁确诊 HZ 的受试者 中未证实预防 PHN 的临床获益。 申请人虽未提供在≥70 岁确诊 HZ 的受试者 中预防 PHN 的 VE,但审评分析认为其结论与在≥50 岁确诊HZ 的受试者一致。 这可能与临床研究中收集到的 PHN 病例数较少,未达到统计学的最低要求有 关。审评认为,PHN 作为 HZ 的并发症,本品在总人群中对 PHN 的预防作用 可归因于预防HZ 的VE,不应将 PHN 单列为适应症。
② 免疫原性方面:
对 ZOSTER-006 研究中 2134 例≥50 岁的免疫原性子集受试者接种前以及 接种后第 3 、14 、26 和 38 个月血清抗 gE 以及抗 VZV 抗体水平进行分析,结 果显示在第 2 剂接种后一个月抗 gE 与 VZV 抗体达到较高水平,直至第 2 剂 接种后三年(第 38 个月)抗体仍维持较高水平; 同时观察到 gE 与VZV 特异 性 CD4+ T 细胞频数增长。ZOSTER-022 研究未评价细胞免疫水平,在 798 例 ≥70 岁的免疫原性子集受试者中观察到抗 gE 以及抗 VZV 抗体水平趋势与 ZOSTER-006 研究结果一致。
将来自 ZOSTER-006 和 022 研究、接种 2 剂疫苗的所有可评估的免疫原 性子集受试者以及所有 HZ 确诊病例(表 )的第 3 个月体液免疫应答与 HZ VE 进行相关性分析(CoP)。验证保护作用关联因素的统计方法采用Prentice 法,识别阈值的统计方法采用 Dunning 回归。
表 6. 用于 COP 分析的受试者
| 队列 | 总计 | ZOSTER-006 | ZOSTER-022 | ||
| 本品 | 安慰剂 | 本品 | 安慰剂 | ||
| CoP 人群-免疫原性亚组 | 3163 | 1151 | 1157 | 422 | 433 |
| CoP 人群-未纳入免疫原性亚组的 HZ 病例 | 446 | 5 | 208 | 23 | 210 |
ZOSTER-006 、ZOSTER-022 第 3 个月抗 gE 抗体浓度的反向累计曲线详 见图 1。描述性数据表明疫苗组与安慰剂组间的抗 gE 抗体浓度分布差异显著,
两组中均存在 HZ 病例的抗 gE 抗体浓度较低的趋势。
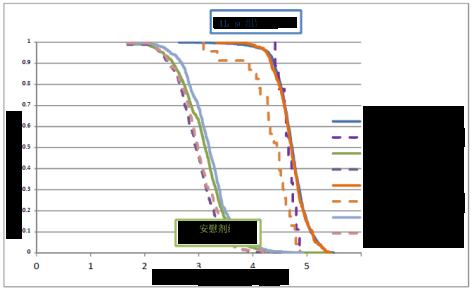
图 1. CoP 人群按研究和临床结局(确诊 HZ 和未确诊 HZ 病例的受试者)划 分后的第 3 个月 gE ELISA 浓度(log10 mIU/mL)的反向累积曲线(RCC)
注:ZOSTER-006 = ZOE 50;ZOSTER-022 =ZOE 70 所有确诊的 HZ 病例基线时血清应答均呈阳性。
在评价抗 gE 抗体浓度与保护作用关联因素时,ZOSTER-022 研究符合 4 条 Prentice 准则。ZOSTER-006 研究和 ZOSTER-006/022 的汇总分析,符合 Prentice 准则的 1~3 条,但不符合第 4 条。在评价抗 gE 抗体自基线水平增加 倍数与保护作用关联因素时,ZOSTER-006 、ZOSTER-022 和两项研究的汇总 分析均不符合任何一条 Prentice 准则。申请人认为 CoP 的概念不需要引入保 护阈值,将抗 gE 抗体浓度指标二分为受保护和未受保护可能导致相关信息丢 失。因此,建议使用基于模型的概率( Dunning 模型)来预测疫苗效力而不 是固定阈值。Dunning 曲线估值预测 VE 的能力详见图 2,如 4 log10 IU/mL 的 阈值对应 84.6 %的保护概率。通过 Dunning 曲线, 申请人根据 ZOSTER-026 研究中接种第 2 剂后观测到的抗 gE ELISA 浓度预测了 0-6 月和 0-2 月免疫程 序的保护效力:0-2 月程序 VE 为 93.1%(95%CI:90.8 – 95.3 );0-6 月为 92.0% (95%CI:89.6 – 94.4)。这一估计与 ZOSTER-006 和 ZOSTER-022 研究中观
测到的 VE 值(91.4%)接近。因此, 申请人认为支持在≥50 岁的受试者中使 用 Dunning 曲线估值预测 HZ VE。
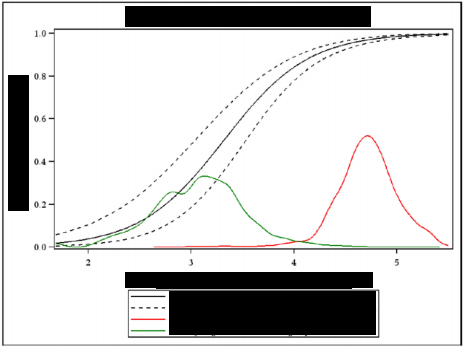
图 2. 抗 gE 抗体浓度预测的保护概率及抗 gE 抗体的 GMC (log 10 IU/mL)
鉴于在 CoP 分析中,gE 抗体水平不能完全满足经典 Prentice 的四项准则, 且不能使用样本量较小和/或模型失拟的理由给出满意解释,故明确第 2 剂疫 苗接种后的 gE 抗体(lg 值)与保护概率关系仅供参考,不支持用于推断保护 效力;采用新的 Dunning 模型的合理性尚待论证。审评认为 gE 抗体暂不宜作 为推断保护效力的生物标志物。
③ 批间一致性方面:
ZOSTER-007 研究入选 651 例受试者,622 例被纳入免疫原性 ATP 人群。 第 2 剂接种后1 个月,本品三个批次经两两比较,抗 gE 抗体 GMC 比值的 95% CI 均在[0.67,1.5]之内;抗 gE 抗体阳性率差值的双侧 95% CI 均在[-10%;+ 10%]之内。
④ 免疫程序探索方面:
ZOSTER-026 研究入选 354 例受试者,343 例受试者被纳入免疫原性 ATP
人群。本品采用 0-6 月、0-12 月接种程序与0-2 月免疫程序比较,第 2 剂接种 后 1 个月抗 gE 抗体阳转率 97.5%CI 下限均高于 60%。0-6 月与 0-2 月免疫程 序相比,抗 gE 抗体校正 GMC 比值 97.5%CI 上限小于 1.5,满足非劣效假设; 0-12 月与 0-2 月免疫程序相比,GMC 比值 97.5% CI 上限大于 1.5,未证实非 劣效。
综合考虑 ZOSTER-006/022 汇总分析的CoP 分析结果及本研究中0-6 月接 种程序与0-2 月接种程序的免疫原性非劣效成立,结合老年人群的特点,为便 于临床应用,审评认可本品第 2 剂在第 1 剂后2~6 个月之间接种。
(3 )亚洲人群有效性数据
在全球范围内,VZV 分离株已经基于序列分析识别了 5 个不同的进化枝。 进化枝分布存在地理差异:在欧洲,进化枝 1(50%)和进化枝 3(37%)占 主导地位;在非洲进化枝 5( 100%),在大洋洲进化枝 1(52%)和进化枝 3 (22%),在美国进化枝 1(70%),在亚洲进化枝 2(74%)占主导地位。
地理差异可能归因于环境因素、宿主-病毒相互作用、选择性进化压力和/或 进口。来自东亚的证据表明,进化枝 2 在整个地区占主导地位。目前最广泛 的研究纳入了分别代表中国大陆的东部、南部、西部、北部和中部的八个省 的 77 份水痘和 11 份带状疱疹样本。98%的水痘和带状疱疹分离株属于进化枝 2,<2%的样本归类为进化枝 4 和进化枝 5。申请人认为尽管 VZV 分型存在 区域差异,但在 VZV 感染中发挥着重要作用的 gE 在所有进化枝分型中都是 高度保守的,氨基酸序列的同源性高于 99%。理论上本品有效性不受地理因 素和 gE 遗传多样性的影响,即预期在中国≥50 岁成人中的有效性类似全球临 床数据。
2019 年 2 月 21~22 日召开的专家咨询会上,流行病学专家认为,尽管 VZV 进化枝的全球分布存在地理差异; 中国地域辽阔,各地间也存在多样性。 但日本等地区的进化枝 2 与中国大多数地区进化枝分布一致。审评认为, 申
请人所引用的中国地区数据仅来源于少量局部流行病学调查研究文献,总体 上中国大陆地区缺少全面详实的带状疱疹相关流行病学及 VZV 、gE 分型数据。 考虑到中国幅员辽阔,各地间可能存在地区差异,建议本品在后续研究中观 察和收集相关数据。
根据 ZOSTER-006 、022 两项关键研究的 EOS 数据生成桥接数据,其中 亚洲地区(中国台湾和中国香港、韩国和日本) 共入组 5196 例受试者,5193 例进入 TVC 人群(表7)。ZOSTER-006/022 汇总研究的 TVC Asian 和 mTVC Asian 的人口统计学特征相似; 除族裔和地理祖先外, 与整个 ZOSTER- 006/022 汇总研究的 TVC 人口统计学特征也相似。
表 7. 亚洲各中心受试者人数(TVC,EOS 分析)
| 国家/地区 | 试验组受试者( %) | 安慰剂组受试者( %) | |
| Zoster-006 | 中国香港 | 236(3.1%) | 234(3.0%) |
| 日本 | 288(3.7%) | 289(3.7%) | |
| 韩国 | 268(3.5%) | 271(3.5%) | |
| 中国台湾 | 640(8.3%) | 640(8.3% | |
| 亚洲合计 | 1432 | 1434 | |
| Zoster -022 | 中国香港 | 91( 1.3%) | 89( 1.3%) |
| 日本 | 256(3.7%) | 255(3.7%) | |
| 韩国 | 263(3.8%) | 265(3.8%) | |
| 中国台湾 | 554(8.0%) | 554(8.0%) | |
| 亚洲合计 | 1164 | 1163 | |
| 合计 | 2596 | 2597 |
在≥50 岁的亚洲亚组受试者中预防 HZ 的 VE 为 95.55%( 95% CI:86.42 – 99.11 ),与整个 ZOSTER-006 研究人群中观察到的疫苗效力相似。在≥70 岁的 亚洲亚组受试者中预防 HZ 的 VE 为 95.26%(95%CI:85.49 – 99.05),与整个 ZOSTER-022 研究人群中观察到的疫苗效力相似。
ZOSTER-006/022 研究汇总分析,亚洲亚组受试者中共收集到 79 例确诊 HZ 的病例,其中,试验组和安慰剂组中分别 4 例、75 例。亚洲亚组受试者中 试验组和安慰剂组的 HZ 总发生率分别为 0.8 和 14.7/1000 人-年。本品在 ZOSTER-006/022 汇总的 mTVC Asian≥70 岁受试者中预防 HZ 的VE 为 94.71%
(95%CI:85.89 – 98.60),与整个 ZOSTER-006/022 汇总研究人群中预防 HZ 的 VE 相似。
亚洲亚组与全人群保护效力的一致性分析:不同地区以及参与研究的各
亚洲国家或地区间,本品对 HZ VE 的点估计值范围为 90.95%至 100%(表、 表 9),95% CI 大部分重叠。Logistic 回归模型评价疫苗效果与国家/地区之间
的相互作用的似然比检验的 p 值远高于 0.05(表 10)。因此, 没有统计证据 表明疫苗接种与国家/地区之间存在显著的相互作用, 即本品预防带状疱疹的 保护力在全球范围内具有一致性。由于报告的 PHN 病例数量有限和提供的信 息数据有限,因此无法解释 PHN 保护力的所有子分析(按整体和按年龄层) 的一致性。
申请人对亚洲人群带状疱疹和 PHN 保护力的评价是基于将两个研究≥50 岁的人群直接合并。审评认为申请人在临床试验方案中对全人群带状疱疹保 护力的评价是基于 Zoster-006 (≥50 岁)和 Zoster-022 (≥70 岁)的单独评价,
因此亚洲人群带状疱疹保护力也应基于 Zoster-006 ( ≥50 岁)和 Zoster-022 ( ≥70 岁) 的单独评价。审评对带状疱疹保护力的区域一致性分析显示,
Zoster-006 研究中,在亚洲≥50 岁人群中,带状疱疹保护力试验组相对对照组 的保护效力为 95.55% (86.42%,99.11%)。Zoster-022 研究,在亚洲≥70 岁人群 中,带状疱疹保护力试验组相对对照组的保护效力为 95.26% (85.49%,99.05%)。
表 8. 采用 Poisson 法按地区划分的事后探索性 HZ VE 分析
(mTVC 人群,≥50 岁,ZOSTER-006/022 汇总分析)
| 本品 | 安慰剂 | VE | 95% CI | |||||||||
| 地区 | N | n | T(年) | n/T (每1000) | N | n | T (年) | n/T (每1000) | (%) | LL | UL | p-值 |
| 澳大利亚* | 343 | 0 | 1303.9 | 0.0 | 351 | 14 | 1272.0 | 11.0 | 100.00 | 76.86 | 100.00 | <0.0001 |
| 欧洲* | 7352 | 14 | 28513.4 | 0.5 | 7432 | 197 | 28317.7 | 7.0 | 92.96 | 87.90 | 96.22 | <0.0001 |
| 拉丁美洲* | 1194 | 4 | 4224.3 | 0.9 | 1217 | 45 | 4272.2 | 10.5 | 91.00 | 75.33 | 97.65 | <0.0001 |
| 北美洲* | 2569 | 8 | 9478.2 | 0.8 | 2572 | 92 | 9294.4 | 9.9 | 91.51 | 82.57 | 96.44 | <0.0001 |
| 亚洲* | 2423 | 6 | 9603.1 | 0.6 | 2463 | 129 | 9471.0 | 13.6 | 95.41 | 89.73 | 98.35 | <0.0001 |
| 总体* | 13881 | 32 | 53122.9 | 0.6 | 14035 | 477 | 52627.2 | 9.1 | 93.36 | 90.50 | 95.51 | <0.0001 |
*按年龄层校正的 VE;p 值=双侧精确P 值取决于病例数
表 9. 采用Poisson 法按亚洲国家/地区划分的 HZ VE 分析
(亚洲亚组 mTVC 人群,≥50 岁,ZOSTER-006/022 汇总分析)
| 本品 | 安慰剂 | VE | 95% CI | |||||||||
| 地区 | N | n | T(年) | n/T (每1000) | N | n | T(年) | n/T (每1000) | ( %) | LL | UL | p-值 |
| 中国香港* | 307 | 0 | 1228.4 | 0.0 | 305 | 16 | 1186.7 | 13.5 | 100.00 | 80.22 | 100.00 | <0.0001 |
| 日本* | 513 | 3 | 2052.8 | 1.5 | 529 | 33 | 2047.0 | 16.1 | 90.95 | 71.15 | 98.22 | <0.0001 |
| 韩国* | 489 | 1 | 1815.2 | 0.6 | 494 | 19 | 1778.3 | 10.7 | 94.88 | 67.76 | 99.88 | <0.0001 |
| 中国台湾* | 1114 | 2 | 4506.7 | 0.4 | 1135 | 61 | 4458.9 | 13.7 | 96.76 | 87.77 | 99.62 | <0.0001 |
| 亚洲* | 2423 | 6 | 9603.1 | 0.6 | 2463 | 129 | 9471.0 | 13.6 | 95.41 | 89.73 | 98.35 | <0.0001 |
*按年龄层校正的 VE;p 值=双侧精确P 值取决于病例数
表 10. 接种(疫苗/安慰剂)与地区或亚洲国家/地区间的相互作用的评价总结
(亚洲亚组 mTVC 人群, ≥50 岁, ZOSTER-006/022 汇总分析)
| 事件类型 | p-值 | |
| 地区vs接种(总体年龄) | HZ | 0.4798 |
| PHN | 0.7107 | |
| 亚洲国家/地区vs接种(总体年龄) | HZ | 0.4221 |
| PHN | 0.7231 | |
| 地区vs接种(50-59岁) | HZ | 0.6635 |
| PHN | NA | |
| 亚洲国家/地区vs接种(50-59岁) | HZ | 0.6644 |
| PHN | NA | |
| 地区vs接种(60-69岁) | HZ | NA |
| PHN | NA | |
| 亚洲国家/地区vs接种(60-69岁) | HZ | 0.4883 |
| PHN | NA | |
| 地区vs接种(≥70岁) | HZ | 0.5995 |
| PHN | NA | |
| 亚洲国家/地区vs接种(≥70岁) | HZ | 0.5726 |
| PHN | NA |
NA=算法因极少数事件而未收敛(Hessian 否定而不是正定)且未进行检验
鉴于对本品的评价主要基于其保护效力,且目前获得的免疫原性结果尚 不能与保护性建立对应关系,审评对亚洲亚组人群的免疫原性结果不做进一 步评价。
主要安全性汇总分析所纳入的 ZOSTER-006 、022 两项研究 TVC 人群的 人口统计学特征,包括平均年龄、种族和地理血统特征在试验组和安慰剂组
之间类似。两项研究共有 29305 例受试者纳入安全性分析集,其中 14645 例 受试者接种了至少 1 剂试验疫苗;4193 例退出研究,其中 1405 例因 SAE 退 出,110 例受试者因非 SAE 退出。4 例因存在因果关系的 SAE 退出研究,其 中试验疫苗组、安慰剂组分别 3 例和 1 例;48 例因存在因果关系的非 SAE 退 出,其中试验疫苗组、安慰剂组分别 44 例、4 例。
审评认为,虽然试验组有 3 例受试者因存在因果关系的 SAE 退出,安慰 剂组仅 1 例,但其在所有接种的受试者中所占比例均较小,无法得出两组间 存在显著差异的结论。试验组因存在因果关系的非 SAE 退出的发生率也高于 安慰剂组,可能与本品的反应原性有关,提示有必要在上市后的临床研究中 继续关注本品的安全性。
申请人选取其中 9946 例受试者(试验组与安慰剂组 1:1 ),作为 7 天日记 卡子集,进行了征集性 AE 的观察。
对于 7 天日记卡子集:疫苗接种后 7 天内,试验组和安慰剂组受试者至 少发生 1 次 AE 的总体发生率分别为 83.9%和 37.4%,至少发生 1 次 3 级 AE
的发生率分别为 16.5%和 3.2%(表)。试验组总体 AE 发生率未见随接种剂次 增加而明显增加的趋势。
表 4. 每剂和所有剂次接种后 7 天内征集和非征集性 AE 发生情况
(7 天日记卡子集 TVC 人群)
| 征集和非征集性AE | ||||||||||||||||
| 任意症状 | 全身症状 | 局部症状 | ||||||||||||||
| 95%CI | 95%CI | 95%CI | ||||||||||||||
| Group | N | n | % | LL | UL | N | n | % | LL | UL | N | n | % | LL | UL | |
| 首剂 | 本品 | 4969 | 3835 | 77.2 | 76.0 | 78.3 | 4969 | 2550 | 51.3 | 49.9 | 52.7 | 4969 | 3584 | 72.1 | 70.9 | 73.4 |
| 安慰剂 | 4977 | 1417 | 28.5 | 27.2 | 29.7 | 4977 | 1232 | 24.8 | 23.6 | 26.0 | 4977 | 426 | 8.6 | 7.8 | 9.4 | |
| 第2剂 | 本品 | 4746 | 3570 | 75.2 | 74.0 | 76.4 | 4746 | 2541 | 53.5 | 52.1 | 55.0 | 4746 | 3268 | 68.9 | 67.5 | 70.2 |
| 安慰剂 | 4773 | 1063 | 22.3 | 21.1 | 23.5 | 4773 | 920 | 19.3 | 18.2 | 20.4 | 4773 | 305 | 6.4 | 5.7 | 7.1 | |
| 总体/剂次 | 本品 | 9715 | 7405 | 76.2 | 75.4 | 77.1 | 9715 | 5091 | 52.4 | 51.4 | 53.4 | 9715 | 6852 | 70.5 | 69.6 | 71.4 |
| 安慰剂 | 9750 | 2480 | 25.4 | 24.6 | 26.3 | 9750 | 2152 | 22.1 | 21.3 | 22.9 | 9750 | 731 | 7.5 | 7.0 | 8.0 | |
| 总体/受试者 | 本品 | 4969 | 4170 | 83.9 | 82.9 | 84.9 | 4969 | 3274 | 65.9 | 64.6 | 67.2 | 4969 | 3953 | 79.6 | 78.4 | 80.7 |
| 安慰剂 | 4977 | 1861 | 37.4 | 36.0 | 38.8 | 4977 | 1648 | 33.1 | 31.8 | 34.4 | 4977 | 591 | 11.9 | 11.0 | 12.8 | |
| 3级征集和非征集性AE | ||||||||||||||||
| 本品 | 4969 | 459 | 9.2 | 8.4 | 10.1 | 4969 | 281 | 5.7 | 5.0 | 6.3 | 4969 | 269 | 5.4 | 4.8 | 6.1 | |
| 首剂 | 安慰剂 | 4977 | 95 | 1.9 | 1.5 | 2.3 | 4977 | 92 | 1.8 | 1.5 | 2.3 | 4977 | 8 | 0.2 | 0.1 | 0.3 |
| 第2剂 | 本品 | 4746 | 529 | 11.1 | 10.3 | 12.1 | 4746 | 382 | 8.0 | 7.3 | 8.9 | 4746 | 270 | 5.7 | 5.0 | 6.4 |
| 安慰剂 | 4773 | 80 | 1.7 | 1.3 | 2.1 | 4773 | 74 | 1.6 | 1.2 | 1.9 | 4773 | 10 | 0.2 | 0.1 | 0.4 | |
| 总体/剂次 | 本品 | 9715 | 988 | 10.2 | 9.6 | 10.8 | 9715 | 663 | 6.8 | 6.3 | 7.3 | 9715 | 539 | 5.5 | 5.1 | 6.0 |
| 安慰剂 | 9750 | 175 | 1.8 | 1.5 | 2.1 | 9750 | 166 | 1.7 | 1.5 | 2.0 | 9750 | 18 | 0.2 | 0.1 | 0.3 | |
| 总体/受试者 | 本品 | 4969 | 822 | 16.5 | 15.5 | 17.6 | 4969 | 573 | 11.5 | 10.7 | 12.5 | 4969 | 460 | 9.3 | 8.5 | 10.1 |
| 安慰剂 | 4977 | 159 | 3.2 | 2.7 | 3.7 | 4977 | 150 | 3.0 | 2.6 | 3.5 | 4977 | 18 | 0.4 | 0.2 | 0.6 |
疫苗接种后 7 天内,试验组和安慰剂组受试者至少 1 种征集性症状的发 生率分别为 84.5%和 33.7%,至少 1 种 3 级征集性 AE 的发生率分别为 16.0% 和 2.5%。与疫苗相关的 AE 按症状分析,最常见的接种部位反应包括:疼痛 (78.0%)、发红(38.1%)和肿胀(25.9%),其中 3 级 AE 发生率分别为 6.4%、 2.9% 、1.0%(表);最常见的全身反应包括:肌痛(40.0%)、疲乏(38.9%)、 头痛(32.6%)、寒战(23.5%)、发热( 17.9%)、胃肠道症状( 13.0%),其中 3 级 AE 发生率由高到低依次为肌痛(4.5%)、疲乏(4.5%)、寒战(3.7%)、
头痛(2.9%)、胃肠道症状( 1.0%)、发热(0.3%)(表)。按接种剂次分析, 接种部位征集性AE 各症状发生率未见随接种剂次增加而显著增加的趋势;全 身征集性 AE 各症状发生率随接种剂次增加稍有增加趋势(未见统计学比较)。 按年龄分层分析,无论是试验组还是安慰剂组,均可见≥70 岁受试者的总体 AE 及 3 级 AE 发生率低于 50-69 岁受试者。
表 5.每剂和所有剂次接种后 7 天内接种部位征集性 AE 按症状分析
(7 天日记卡子集 TVC 人群)
| 本品 | 安慰剂 | ||||||||||
| 95%CI | 95%CI | ||||||||||
| 症状 | 类型 | N | n | % | LL | UL | N | n | % | LL | UL |
| 首剂 | |||||||||||
| 疼痛 | 所有 | 4861 | 3417 | 70.3 | 69.0 | 71.6 | 4865 | 378 | 7.8 | 7.0 | 8.6 |
| 3级 | 4861 | 171 | 3.5 | 3.0 | 4.1 | 4865 | 7 | 0.1 | 0.1 | 0.3 | |
| 发红(mm) | 所有 | 4861 | 1368 | 28.1 | 26.9 | 29.4 | 4865 | 43 | 0.9 | 0.6 | 1.2 |
| >100 | 4861 | 91 | 1.9 | 1.5 | 2.3 | 4865 | 0 | 0.0 | 0.0 | 0.1 | |
| 肿胀(mm) | 所有 | 4861 | 895 | 18.4 | 17.3 | 19.5 | 4865 | 33 | 0.7 | 0.5 | 1.0 |
| >100 | 4861 | 30 | 0.6 | 0.4 | 0.9 | 4865 | 0 | 0.0 | 0.0 | 0.1 | |
| 第2剂 | |||||||||||
| 疼痛 | 所有 | 4699 | 3089 | 65.7 | 64.4 | 67.1 | 4715 | 281 | 6.0 | 5.3 | 6.7 |
| 3级 | 4699 | 197 | 4.2 | 3.6 | 4.8 | 4715 | 10 | 0.2 | 0.1 | 0.4 | |
| 发红(mm) | 所有 | 4699 | 1288 | 27.4 | 26.1 | 28.7 | 4715 | 27 | 0.6 | 0.4 | 0.8 |
| >100 | 4699 | 71 | 1.5 | 1.2 | 1.9 | 4715 | 0 | 0.0 | 0.0 | 0.1 | |
| 肿胀(mm) | 所有 | 4699 | 830 | 17.7 | 16.6 | 18.8 | 4715 | 17 | 0.4 | 0.2 | 0.6 |
| >100 | 4699 | 25 | 0.5 | 0.3 | 0.8 | 4715 | 0 | 0.0 | 0.0 | 0.1 | |
| 总体/受试者 | |||||||||||
| 疼痛 | 所有 | 4884 | 3810 | 78.0 | 76.8 | 79.2 | 4880 | 533 | 10.9 | 10.1 | 11.8 |
| 3级 | 4884 | 315 | 6.4 | 5.8 | 7.2 | 4880 | 17 | 0.3 | 0.2 | 0.6 | |
| 发红(mm) | 所有 | 4884 | 1863 | 38.1 | 36.8 | 39.5 | 4880 | 64 | 1.3 | 1.0 | 1.7 |
| >100 | 4884 | 141 | 2.9 | 2.4 | 3.4 | 4880 | 0 | 0.0 | 0.0 | 0.1 | |
| 肿胀(mm) | 所有 | 4884 | 1267 | 25.9 | 24.7 | 27.2 | 4880 | 48 | 1.0 | 0.7 | 1.3 |
| >100 | 4884 | 51 | 1.0 | 0.8 | 1.4 | 4880 | 0 | 0.0 | 0.0 | 0.1 | |
表 6. 所有剂次接种后 7 天内全身征集性 AE 按症状分析
(7 天日记卡子集 TVC 人群)
| 本品 | 安慰剂 | ||||||||||
| 95%CI | 95%CI | ||||||||||
| 症状 | 类型 | N | n | % | LL | UL | N | n | % | LL | UL |
| 疲乏 | 所有 | 4876 | 2172 | 44.5 | 43.1 | 46.0 | 4881 | 805 | 16.5 | 15.5 | 17.6 |
| 3级 | 4876 | 257 | 5.3 | 4.7 | 5.9 | 4881 | 50 | 1.0 | 0.8 | 1.3 | |
| 相关 | 4876 | 1895 | 38.9 | 37.5 | 40.2 | 4881 | 575 | 11.8 | 10.9 | 12.7 | |
| 相关的3级 | 4876 | 218 | 4.5 | 3.9 | 5.1 | 4881 | 38 | 0.8 | 0.6 | 1.1 | |
| 胃肠道症状 | 所有 | 4876 | 842 | 17.3 | 16.2 | 18.4 | 4881 | 426 | 8.7 | 8.0 | 9.6 |
| 3级 | 4876 | 66 | 1.4 | 1.0 | 1.7 | 4881 | 27 | 0.6 | 0.4 | 0.8 | |
| 相关 | 4876 | 636 | 13.0 | 12.1 | 14.0 | 4881 | 228 | 4.7 | 4.1 | 5.3 | |
| 相关的3级 | 4876 | 48 | 1.0 | 0.7 | 1.3 | 4881 | 12 | 0.2 | 0.1 | 0.4 | |
| 头痛 | 所有 | 4876 | 1838 | 37.7 | 36.3 | 39.1 | 4881 | 755 | 15.5 | 14.5 | 16.5 |
| 3级 | 4876 | 162 | 3.3 | 2.8 | 3.9 | 4881 | 34 | 0.7 | 0.5 | 1.0 | |
| 相关 | 4876 | 1588 | 32.6 | 31.3 | 33.9 | 4881 | 511 | 10.5 | 9.6 | 11.4 | |
| 相关的3级 | 4876 | 143 | 2.9 | 2.5 | 3.4 | 4881 | 21 | 0.4 | 0.3 | 0.7 | |
| 肌痛 | 所有 | 4876 | 2180 | 44.7 | 43.3 | 46.1 | 4881 | 570 | 11.7 | 10.8 | 12.6 |
| 3级 | 4876 | 248 | 5.1 | 4.5 | 5.7 | 4881 | 33 | 0.7 | 0.5 | 0.9 | |
| 相关 | 4876 | 1949 | 40.0 | 38.6 | 41.4 | 4881 | 421 | 8.6 | 7.9 | 9.4 | |
| 相关的3级 | 4876 | 218 | 4.5 | 3.9 | 5.1 | 4881 | 22 | 0.5 | 0.3 | 0.7 | |
| 寒战 | 所有 | 4876 | 1307 | 26.8 | 25.6 | 28.1 | 4881 | 281 | 5.8 | 5.1 | 6.4 |
| 3级 | 4876 | 198 | 4.1 | 3.5 | 4.7 | 4881 | 13 | 0.3 | 0.1 | 0.5 | |
| 相关 | 4876 | 1148 | 23.5 | 22.4 | 24.8 | 4881 | 193 | 4.0 | 3.4 | 4.5 | |
| 相关的3级 | 4876 | 181 | 3.7 | 3.2 | 4.3 | 4881 | 10 | 0.2 | 0.1 | 0.4 | |
| 发热/(*)(°C) | 所有 | 4876 | 1002 | 20.5 | 19.4 | 21.7 | 4881 | 145 | 3.0 | 2.5 | 3.5 |
| [37.5-38.1] | 4876 | 701 | 14.4 | 13.4 | 15.4 | 4881 | 111 | 2.3 | 1.9 | 2.7 | |
| [38.1-39.1] | 4876 | 276 | 5.7 | 5.0 | 6.3 | 4881 | 21 | 0.4 | 0.3 | 0.7 | |
| >39.0 | 4876 | 14 | 0.3 | 0.2 | 0.5 | 4881 | 8 | 0.2 | 0.1 | 0.3 | |
| 相关 | 4876 | 872 | 17.9 | 16.8 | 19.0 | 4881 | 84 | 1.7 | 1.4 | 2.1 | |
| >39.0相关 | 4876 | 13 | 0.3 | 0.1 | 0.5 | 4881 | 2 | 0.0 | 0.0 | 0.1 | |
接种后 30 天内,试验组、安慰剂组与疫苗接种相关的非征集 AE 发生率 分别为 6.5% 、2.8%,其中 3 级 AE 发生率分别为 0.7% 、0.2%。最常报告的与 疫苗接种相关非征集AE 为注射部位瘙痒,试验组和安慰剂组分别为发生率分 别为 1.1 % 、0.1%。
对于 TVC 人群:接种后 30 天内,试验组、安慰剂组非征集性 AE 的发生 率分别为 50.5% 、32.0%,其中 3 级 AE 发生率分别为 7.5% 、3.8%。试验组报 告的非征集性 AE 明显高于安慰剂组。上述不平衡的主要原因是,未包含在 7
天日记卡子集中的受试者将预期的局部症状和全身症状报告为非征集性 AE。
与安慰剂组相比,试验组发生率较高的大多数非征集性 AE 属于“全身性疾病 及接种部位反应”的系统器官分类(SOC),且与 7 天日记卡子集人群的接种 后 7 天内征集性 AE 相重合。下述四种非征集性 AE,试验组受试者发生率 ≥1.0%且至少为安慰剂组发生率的2 倍:( 1 )注射部位瘙痒:试验组2.16% , 安慰剂组 0.24%;RR=9.07[95%CI:6.38-13.25],未校正 P值<0.00001;(2) 不适:试验组 1.73%,安慰剂组 0.29%;RR=5.91[95%CI:4.27-8.37],未校正 P 值<0.00001;( 3 )疼痛:试验组 1.39%,安慰剂组 0.23%;RR=6.01[95%CI: 4.16-8.91],未校正 P 值<0.00001;(4)注射部位发热:试验组 1.02%,安慰 剂组 0.03%;RR=29.83[95%CI: 12.50-93.22],未校正 P 值<0.00001。综上, 试验组高于安慰剂组的非征集性 AE 大多符合疫苗一般反应原性且均为3 级以 下。
审评还关注到以下症状在两组间发生率有较大差异,建议在后续国内上 市后研究中进一步关注:( 1 )注射部位蜂窝织炎【试验组 6 例(0.04%)、 安慰剂组 0 例(0.00%)】;(2)痛风【试验组 26 例(0.18%)、安慰剂组 7 例(0.05%)】; (3 )关节痛【试验组 252 例( 1.72%)、安慰剂组 171 例 ( 1.17%)】。审评未见上述病例详细信息, 但对于痛风, 申请人提交的 FDA 审评报告中有如下描述, “接种后 30 天内,试验组中 26 例受试者和安慰 剂组中 7 例受试者报告了痛风;各组均有 1 例受试者报告了痛风性关节炎。 该时间段报告痛风的受试者中,试验组中 19 例受试者和安慰剂组中 3 例受试 者均为首次报告痛风事件” 。痛风已被申请人纳入拟定的主动监测研究。
接种后 30 天内,试验组、安慰剂组与疫苗接种相关的非征集 AE 发生率 分别为 34.5% 、6.6%(968 例),其中 3 级 AE 发生率分别 4.0% 、0.4%。 除了 与 7 天日记卡子集人群的征集性 AE 相同的症状之外,试验组最常报告的与疫 苗接种相关非征集性 AE (≥1.0%的受试者)为注射部位瘙痒(2.1%)、不适
( 1.6%)、疼痛( 1.2%)、注射部位发热( 1.0%)。上述 4 种非征集性 AE 在安慰剂组发生率均≤0.2%。
审评认为,本品是含 AS01B 佐剂系统的新疫苗, 目前仅在 7 天日记卡子 集中采用日记卡方式进行征集性症状监测,对于非 7 天日记卡组受试者将接 种后所有局部和全身症状均报告为非征集性 AE,导致总接种人群接种后 30 天内非征集性 AE 中很大比例来自上述非7 天日记卡组受试者接种后7 天内局 部和全身症状。 因此,审评要求对于 7 天日记卡子集和 TVC 人群应分别汇总 接种后 30 天内的整体安全性数据。鉴于该项分析仅涉及安全性数据的表述方 式,故可在上市后进行。
整个接种后随访期内,试验组与安慰剂组各报告了 15 例与疫苗接种相关 的 SAE,包括: 昏厥、风湿性关节炎、血小板减少性紫癜等。 大多数与疫苗 接种相关 SAE 报告于最后一剂接种后 1 年内(试验组 15 例、安慰剂组 13 例);而从首剂接种至最后一剂接种后 30 天,试验组和安慰剂组各有 8 例受 试者报告了相关 SAE。致命性 SAE 中,1 例中性粒细胞减少性脓毒症被研究 者认为与疫苗接种有关。该事件报告 ZOSTER-022 中试验组的 1 例 90 岁男性 受试者,在接种首剂本品后 75 天出现严重的 3 级急性髓系白血病(AML),住 院治疗并退出研究。因中性粒细胞减少性脓毒症,该受试者在接种首剂本品 后 97 天死亡。研究者认为, 急性髓系白血病和中性粒细胞减少性脓毒症均可 能由本品接种引起, 同时也可能与该受试者的临床治疗有关(急性髓系白血 病的化疗、特发性血小板减少症的长期治疗)。
在整个接种后随访期内,试验组 179 例( 1.2%)、安慰剂组 202 例( 1.4%) 受试者报告潜在免疫介导性疾病(pIMD)。 约半数 pIMD 发生于最后一剂接 种后 1 年以上。两组最常报告的 pIMD (≥10 例受试者)按 PT 分类为风湿性 多肌痛(试验组 32 例,安慰剂组 29 例)、类风湿性关节炎(试验组 20 例,安 慰剂组 26 例)、银屑病(试验组 15 例,安慰剂组 18 例)和自身免疫性甲状腺
炎(试验组 13 例,安慰剂组 10 例)。总体而言,两组未见显著差异。其中, 试验组 16 例(0.1%)、安慰剂组 18 例(0.1%)受试者报告的 pIMD 被研究 者认为与疫苗接种相关。按照 PT 分类,除风湿性多肌痛(试验组 1 例,安慰 剂组 3 例)、类风湿性关节炎(每组各 2 例)、干燥综合征(安慰剂组 2 例) 和银屑病(试验组 2 例,安慰剂组 1 例)外,大多数相关 pIMD 每组至多 1 例 受试者报告。按年龄层分析,50-69 岁和≥70 岁年龄层中,首剂接种至最后一 剂接种后 30 天和 1 年内及整个接种后随访期内报告 pIMD 的受试者发生率类 似。
关于 15 例与疫苗接种相关的 SAE 、16 例与疫苗接种相关的 pIMD,考虑 到相关性判断在揭盲前进行,且试验组与对照组 SAE 、pIMD 的发生率无显著 差异,审评基本认可本品安全性。
若不考虑接种相关性,审评还关注到以下两组间的差异:过敏性血管炎 共报告 2 例,均在试验组;颞动脉炎共报告 9 例(试验组 6 例,安慰剂组 3 例)。白细胞破碎性血管炎、缺血性视神经病变和颞动脉炎已被申请人纳入 拟定的主动监测研究。
对于亚洲人群:总体而言,亚洲亚组中的征集性局部症状、征集性全身 症状、非征集性 AE 、SAE 以及pIMD 与总体研究的结果相似。但在ZOSTER- 006 、ZOSTER-022 以及 ZOSTER-006/022 汇总分析结果中, 任何级别的非征 集性AE、与疫苗接种相关的非征集性AE 以及需参加医疗访视的非征集性AE, TVC 亚洲试验组的发生率均高于 TVC 试验组。基于申请人提交的亚洲人群有 效性和安全性分析结果,建议本品有条件批准上市后在国内开展临床试验,
进一步考察本品在国内人群中的保护效力和安全性。
申请人按CTD格式提交了在欧盟注册的全套申请资料:临床部分涉及M2、
M5两个模块,同时提交了亚洲亚组人群的统计分析数据以及说明书样稿。药 学部分主要分为按照ICH要求提交的NDA CTD资料、上市后变更(3项)、主 动沟通过程中问题答复等评价部分。统计部分本次递交了该品种相关研究的 临床研究方案、临床总结报告、原始数据库、分析数据库及数据说明文件。 申请人提供的数据为xpt格式,所提交资料基本符合生物统计学审评要求。
生物统计学审评确认了本品在 ZOSTER-006 ( ≥50 岁)、ZOSTER-022 (≥70 岁)研究中的 HZ VE;对 HZ VE 的区域一致性分析结果表明,Zoster- 006 研究以及 Zoster-006/022 汇总研究尚不能认为区域间存在不一致趋势。尽 管 Zoster-022 研究显示区域间存在不一致倾向,但由于非亚洲地区人群也能 得出优效结论,且亚洲人群的 HZ VE 相对更高,审评基本认可亚洲与全人群 HZ VE 的一致性。
4.5.1 已知风险
对本品的安全性评价主要来源于 ZOSTER-006 和 ZOSTER-022,共计 14645 例受试者接种了至少一剂本品,14660 例受试者接种了生理盐水。接种 本品后报告的征集性局部不良反应主要为疼痛、发红和肿胀;征集性全身不 良反应主要为肌痛、疲乏、头痛、寒颤、发热和胃肠道症状。观察到的绝大 多数征集性局部和全身不良反应中位持续时间为 2 至 3 天,可自行缓解。 与 50-69 岁的受试者相比,70 岁及以上受试者的征集性局部和全身症状的发生率 较低。征集性局部反应未见随接种剂次增加的趋势,但第 2 剂接种后受试者 报告头痛和寒颤,3 级头痛、寒颤、肌痛和疲乏的比例高于第 1 剂。
除了与征集性 AE 相同的症状之外,试验组最常报告的 (≥1.0%的受试者) 与疫苗接种相关的非征集性 AE 为注射部位瘙痒、不适、疼痛、注射部位发
热。本品受试者报告的可能与接种存在相关性的 SAE 为淋巴结炎、发热超过 39°C(各 1 例)。
针对临床试验中观察到的不良反应, 已纳入本产品的的说明书,并在今 后的临床试验和上市应用中给予关注和对症处理。
4.5.2 潜在风险
本品临床试验期间未报告与疫苗接种相关的注射部位蜂窝织炎,上市后 使用期间报道的绝对发生率也并不高,但审评发现注射部位蜂窝织炎在两组 间不均衡,建议在上市后研究中进一步关注。
接种本品的受试者中痛风(包括痛风性关节炎) 的发生率高于安慰剂组, 但现有的信息不足以确定与本品接种存在相关性。
本品和安慰剂接种组之间,最常报告的 pIMD (≥10 例)为风湿性多肌痛、 类风湿性关节炎、自身免疫性甲状腺炎和银屑病,两组发生频率相当。 审评 关注到过敏性血管炎、颞动脉炎在两组间不均衡,建议在上市后研究中进一 步关注 pIMD。
在本品上市后的使用过程中, 已确定了不良事件—超敏反应,包括血管 性水肿、皮疹及荨麻疹(免疫系统疾病)。由于这些事件为规模不详人群的 自发报告, 因此不一定能够可靠地估计其发生频率或确定其与疫苗之间的因 果关系。
HZ 是由 VZV 复苏引起的急性感染性疾病,好发于老年人和免疫低下人 群,常伴剧烈的顽固性PHN,严重影响患者生活质量。 此外,HZ 还可导致病 毒播散、中风、脑炎和视力损伤(包括失明) 等严重并发症。HZ 发病率较高, 研究发现 90%以上的成人均感染过 VZV,其中约 20%的血清学阳性个体在一 生中会发生 HZ。HZ 的治疗包括及时应用抗病毒和镇痛类药物,但这些药物
只能减少急性 HZ 患者的不适及疼痛症状,不能明显的缩短 HZ 的病程以及缓 解或者减少后遗症的发生。随着我国社会老龄化加剧,HZ 防控形势更加严峻。 目前国内尚无用于预防带状疱疹的疫苗。
本品临床数据显示在≥50 岁人群中预防 HZ 的有效性在90%以上,且在不 同年龄层中保护效力相当。接种本品后大多数受试者均报告了局部不良反应 (疼痛 、发红和肿胀)和/或全身不良反应(头痛 肌痛、疲乏、头痛、寒颤、 发热和胃肠道症状),但通常持续时间短,可自行缓解。整体上试验组与安 慰剂组的受试者死亡率、SAE 和 pIMDs 报告的比例大致相当。
综合评估本品风险与获益,建议批准本品进口用于≥ 50 岁人群。
审评基本认可申请人提供的境外临床试验数据,建议采用境外数据有条 件批准本品在国内上市,用于≥50 岁人群预防 HZ。
关于 PHN:审评认为 PHN 作为 HZ 的并发症,本品对总体 PHN 的益处 可以归因于预防 HZ 的益处;且本品未在确诊 HZ 的患者中证实预防 PHN 的 临床获益,故不建议将 PHN 单列为适应症。可在说明书中明确“临床研究中 提示本品有预防 PHN 的获益,但由于 PHN 例数有限,本品在确诊 HZ 的患者 中预防 PHN 的效力尚未证实”。
关于上市后研究:考虑到本品境外临床研究存在病例诊断流程缺陷及仅 在 7 天日记卡子集中主动收集征集性 AE 等安全性监测问题; 国内缺少带状疱 疹流行病学数据,无法完全明确国内外 VZV 进化枝间的差异, 以及境外数据 中亚洲亚组人群样本量较小等因素,建议上市后应继续开展规范的临床保护 效力试验, 以验证本品在一定样本量下的中国人群安全性和有效性, 同时继 续探索本品保护机制(CMI 等)。
申请人提交了 EPI-ZOSTER-034 (安 全 性 监 测 PMS 队列研究)、 ZOSTER-076(随机对照的 IV 期临床试验) 两项上市后研究方案摘要。审评 建议将两项临床研究进行合并设计一个完整的保护效力研究方案,参考本品 境外临床试验数据和临床意义,提高中国临床试验假设参数,95CI%下限至 少应>25%; 同时满足安全性观察需要(0.1%-1%不良反应发生率),建议本 品试验组应不低于 3000 例,根据上述要求重新估算样本量,并进一步加强安 全性主动监测访视。申请人重新提交了修改后的上市后研究方案 Zoster-076 (rev)摘要。Zoster-076(rev)与 ZOSTER-076 相比研究设计基本不变,样 本量有所增加;同时纳入 EPI-ZOSTER-034 研究中所收集的安全性信息,所 有受试者将在末次接种后至少随访 1 年。根据计算, 预计入组约 6138 例受试 者,每组 3069 例。审评基本认可 Zoster-076(rev.)研究设计, 鉴于申请人仅 递交了方案摘要, 尚待进一步完善细化可实施的方案。在国内临床研究开展 前, 申请人必要时可依据国家局发布的沟通交流管理规定,与药审中心进行 沟通。
本品境外临床研究仅在 7 天日记卡子集中采用日记卡方式进行征集性症 状监测,对于非 7 天日记卡组受试者将接种后所有局部和全身症状均报告为 非征集性 AE,导致总接种人群接种后30 天内非征集性 AE 中很大比例来自上 述非 7 天日记卡组受试者接种后 7 天内局部和全身症状。 因此,审评要求对 于全球人群与亚洲亚组的7 天日记卡子集和 TVC 人群应分别汇总接种后30 天 内的整体安全性数据,并完善说明书的相关内容。本次说明书【不良反应】 内容起草暂先参考 FDA 说明书格式和内容,按全人群和亚洲亚组人群分别描 述。待本品国内保护效力试验完成后再按国内要求更新说明书此部分内容。
(略)
本品未进行生产现场检查。
本品未进行生产现场检查,因此未进行动态核查过程中的抽样检验。
NA
本品为接受境外数据品种,暂未启动临床试验数据核查。
(1)质量可控性
(略)
(2)安全有效性
本品全球临床数据显示在≥50 岁人群中预防 HZ 的有效性在90%以上,且 在不同年龄层中保护效力相当。接种本品后大多数受试者均报告了局部不良 反应(疼痛、发红和肿胀)和/或全身不良反应(头痛 肌痛、疲乏、头痛、 寒颤、发热和胃肠道症状),但通常持续时间短,可自行缓解。整体上试验 组与安慰剂组的受试者死亡率、SAE 和 pIMDs 报告的比例大致相当。
本品在亚洲亚组人群中的保护效力、安全性与全球人群基本一致。 综合 评估本品风险与获益,建议批准本品进口注册用于≥ 50 岁人群预防 HZ,免疫 程序为两剂,第 2 剂与第 1 剂间隔 2 个月接种。如需改变免疫程序,第 2 剂在 第 1 剂后2~6 个月之间接种。
本品应按要求开展国内保护力研究, 安全性监测重点关注蜂窝织炎、 pIMDs 等;按相关法规要求开展药物警戒,遵照事先制定的风险控制措施实 施,并及时提交本品定期安全性报告。待国内保护效力试验完成后再按国内 要求更新说明书安全性部分内容。
经风险效益评估,支持有条件批准本品进口用于 50 岁及以上成人,预防 带状疱疹,不适用于预防原发性水痘。
临床方面:
1.在国内开展临床保护力研究,其临床研究方案【Zoster-076(rev)】尚 需进一步完善。安全性监测重点关注蜂窝织炎、 pIMDs 等;同时探索本品的 保护机制 (CMI 等)。
2.按相关法规要求开展药物警戒,遵照事先制定的风险控制措施实施, 并及时提交本品定期安全性报告。
3.上市后更新说明书相关内容:( 1 )继续完成本品境外全球临床数据的 安全性数据分析;(2)本品国内临床保护效力研究完成后进一步修订。
药学方面:
(略)
同上市后要求,本品上市后的风险控制措施包括:( 1 )在国内开展临床 保护力研究,安全性监测重点关注蜂窝织炎、pIMDs 等。(2 )按相关法规要
求开展药物警戒,遵照事先制定的风险控制措施实施,并及时提交本品定期 安全性报告。
(略)
Hits: 343