
Contents
- 1 目录
- 2 1. 简介
- 3 2. 潜在的病毒污染源
- 4 2.1 主细胞库可能存在的病毒
- 5 3. 细胞系检定:病毒检测
- 6 4. 未加工收获液的病毒检测
- 7 5. 对纯化后产品进行病毒清除研究和病毒检测的依据和行动计划
- 8 6. 病毒清除工艺的评价和鉴定
- 9 7. 连续生产工艺的考虑要点
- 10 8. 总结
- 11 9. 术语表
- 12 附件 1:已鉴定细胞库经体内培养表达的产品
- 13 附件 2:病毒清除研究用病毒的选择
- 14 附件 3:评估病毒和病毒下降因子的统计学考虑
- 15 附件 4:确定病毒清除率研究中下降因子的计算
- 16 附件 5:每剂量病毒颗粒的估算
- 17 附件 6:应用先验知识(包括内部经验)以减少特定产品验证工作的举例
- 18 附件 7:基因工程病毒载体和病毒载体衍生产品
ICH指导原则《Q5A(R2):来源于人或动物细胞系生物技术产品的病毒安全性评价》现进入第3阶段征求意见。按照ICH相关章程要求,ICH监管机构成员需收集本地区关于第2b阶段指导原则初稿的意见并反馈ICH。
Q5A(R2)原文和中文译文见附件,现就该指导原则内容及中文译文翻译稿向社会公开征求意见。
社会各界如有意见(填写附件3),请于2023年1月20日前通过电子邮箱反馈我中心。
联系人:赛文博、于鹏丽
邮 箱:saiwb@cde.org.cn;yupl@cde.org.cn
附件:1.【英文】Q5A(R2):来源于人或动物细胞系生物技术产品的病毒安全性评价(初稿)
2.【中文】Q5A(R2):来源于人或动物细胞系生物技术产品的病毒安全性评价(初稿)
3.【模板】征求意见反馈表
国家药品监督管理局药品审评中心
2022年11月28日
在 ICH 进程的第 2 阶段,根据国家或地区程序,ICH 专家工作组商定的共识性草案或指导原则,由 ICH 大会转交给 ICH 的地区监管机构,用于内部和外部征求意见。
| 代码 | 历史 | 日期 |
| Q5A | 指导委员会批准作为第二阶段草案发布,对外征求意见。 | 1995 年 12 月 1 日 |
| Q5A | 指导委员会批准作为第四阶段草案推荐给 ICH 三方的管理机构采纳。 | 1997 年 7 月 5 日 |
| 代码 | 历史 | 日期 |
| Q5A(R1) | 指导委员会批准了第四阶段后的编辑修正。 | 1999 年 9 月 23 日 |
| 代码 | 历史 | 日期 |
| Q5A(R2) | 由 ICH 大会成员在第二阶段签署并发表用于征求公众意见。 | 2022 年/9 月/29 日 |
法律声明:本文受版权保护,除 ICH 标志外,在始终承认 ICH 版权的前提下,基于公共许可可以使用、复制、在其他作品中引用、改编、修改、翻译或传播。如果对本文件进行任何改编、修改或翻译,必须使用合理步骤来清晰标注、界定或以其它方式明确对原始文件或基于原始文件所做的更改。必须避免任何暗示 ICH 授权或支持对原始文件的改编、修订或翻译的行为。
本文件“按原样”提供,不提供任何形式的担保。在任何情况下,ICH 或原始文件的作者不对因使用本文件造成的任何索赔、伤害或其他责任负责。
上述许可不适用于第三方提供的内容。因此,对于版权归属第三方的文件,必须从该版权所有者处获得复制许可。
|
1. 简介
本指导原则旨在为生物技术产品提供病毒安全性的测试和评价方法,并概述了此类产品上市申请和注册时应提交的研究资料。生物技术产品包括从人或动物来源的特征细胞库(例如,哺乳动物、鸟类、昆虫)引入的细胞培养物中提取的生物治疗药物和特定生物制品。本文件所述“病毒”不包括非常规性传播因子,如与哺乳动物朊病毒(如疯牛病(BSE)、瘙痒病(scrapie))有关的传播因子。鼓励申请人与相应的监管机构讨论疯牛病的相关问题。
本文件涵盖利用重组 DNA 技术并通过体外细胞培养生产的产品,如干扰素、单克隆抗体和重组亚单位疫苗。它还涵盖了从体内如腹水内培养的杂交瘤细胞中提取的产品:有关体内所繁衍细胞测试的特别注意点和附加资料,见附件 1。本文件还适用于某些可进行病毒清除,而不会对产品产生负面影响的基因工程病毒载体和病毒载体衍生产品。此类产品可能包括使用瞬时转染或从稳定细胞系生成的病毒载体,或使用重组病毒感染生成的病毒载体。还包括病毒载体衍生的重组蛋白,例如,杆状病毒表达的病毒样颗粒(Virus-Like Particles, VLP)、蛋白质亚基和基于纳米颗粒的疫苗和治疗药物。此外,该范围还涵盖依赖于辅助病毒,如杆状病毒、单纯疱疹病毒或腺病毒,进行生产的腺相关病毒(Adeno-Associated Virus,AAV)基因治疗载体。关于基因工程病毒载体和病毒载体衍生产品的具体指南见附件 7。灭活病毒疫苗和含自我复制因子的减毒活病毒疫苗不在本文件的范围内。
来源于细胞系的生物技术产品的一个共同特点是存在病毒污染的风险,这种污染在临床上可产生严重的后果。污染可来自原细胞系(细胞基质)本身,也可来自生产过程中偶然带入的外源病毒。然而,到目前为止从细胞系来源的生物技术产品还没有发生传播病毒的问题。尽管如此,有关这些产品病毒污染的安全性,只有通过实行全面的病毒检测程序并在生产过程中对病毒的去除或灭活作用进行评估,才可能得到确实的保证。控制生物技术产品的潜在病毒污染,可归纳为以下三条互补的原则:
- 选择并对细胞系和其他原材料(包括培养基组分)进行检定,确保其不含非预期感染性病毒;
- 评估生产工艺清除感染性病毒的能力;在生产的适当步骤对产品进行测试,确保产品未受感染性病毒的污染。
基因工程病毒载体和病毒载体衍生产品生产过程中使用的一些病毒清除步骤可能无法达到与重组蛋白生产过程中相同的有效性。在这种情况下,可以考虑进一步降低风险,如处理原材料和进行广泛的病毒检测(见附件 7)。
根据统计原则,定量病毒测定检测低浓度病毒的能力取决于样本量。因此,要确定产品中没有感染性病毒污染物,不能单凭是否能够直接测到污染物而定,还需证明纯化方法是否能去除或灭活病毒。
应在具体案例具体分析和逐步推进的基础上,确定在不同生产步骤中所采用的病毒测试方法和病毒清除研究的类型和程度,。应考虑的因素包括:细胞库鉴定和表征研究程度、所测定病毒的性质、培养基组成、培养方法、设施和设备设计、细胞培养后病毒测试结果、生产工艺清除病毒的能力和产品类型及其预期临床用途。本文件的目的是为病毒测试、评估病毒清除率的试验、设计病毒检测和病毒清除研究的推荐方法提供一个总体框架。
制造商应根据其特定产品及其生产过程调整本文推荐的方案。制造商应对其确保病毒安全性的总体方针作出解释并说明其依据。除了提供详细资料外,还应提供一份病毒安全性评估的全面总结,以帮助和利于管理部门审查。该总结应对病毒安全性研究的各个方面,以及用于预防病毒污染的策略进行简要说明。
2. 潜在的病毒污染源
生物技术产品的病毒污染可来自细胞系的原始来源或来自生产过程(包括重组生产细胞系和/或细胞系建库)中偶然带入的外源病毒。在附件 7 中讨论了从主病毒种子( Master Virus Seed,MVS)或工作病毒种子(Working Virus Seed,WVS)中可能偶然引入潜在的外源病毒。使用充分表征的细胞库和 MVS 或 WVS 可降低病毒污染的风险。此外,用于生产重组蛋白、VLP 或病毒载体基因治疗产品的辅助病毒也被视为工艺相关病毒污染物(见附件7)。
2.1 主细胞库可能存在的病毒
细胞可能存在潜伏性或持续性病毒(如疱疹病毒)和内源性逆转录病毒,并且这些病毒可从一代细胞直接传给下一代细胞。在这种情况下,病毒可能以组成型表达或意外地表达成感染性病毒。
病毒可通过以下多种途径进入主细胞库(MCB),例如:1)从受感染动物制备的细胞系;2)使用病毒建立细胞系;3)使用受污染的生物试剂(例如用于选择的抗体)或细胞培养的原材料(例如动物或人血清和猪胰蛋白酶);4)细胞处理和建库过程中受到的污染。
2.2 生产过程中可能引入的外源病毒
外源病毒可通过多种途径污染生产过程,包括但不限于以下途径:1)在细胞培养期间使用受污染的生物原材料或试剂,如动物血清组分;2)使用病毒或病毒载体(包括其生产中使用的辅助病毒)诱导表达编码目的蛋白的特定基因(见附件 7);3)在下游纯化过程中使用受污染的原材料或试剂,例如用于产品选择或纯化的单克隆抗体偶联亲和树脂;4)在处方配制中使用受污染的辅料;5)来自环境的污染,包括非生物原材料的储存或细胞培养和培养基处理期间所受的污染。
对细胞培养的参数进行监控有助于早期发现潜在的外源病毒污染。制造商应尽可能避免在生产过程中使用人和动物来源的原材料(例如人血清、牛血清、猪胰蛋白酶)。在无法做到这一点的情况下,使用动物来源的原材料应基于相应的风险提供文件或资质证明予以支持。包括诸如原产国、组织来源、材料生产过程中应用的病毒灭活/去除步骤以及对原材料进行的病毒检测类型等信息。
在可能的情况下,细胞培养基或其他基质的补充处理方法,如伽马辐照、病毒过滤、高温短时杀菌或 UV-C 辐照可用作额外的病毒风险缓解措施。
3. 细胞系检定:病毒检测
适当的病毒检测是确定细胞系是否适用于生物技术产品生产的重要部分。
3.1 对主细胞库、工作细胞库及达到体外细胞传代限次的生产用细胞进行病毒检测的建议
不同级别细胞(包括 MCB、WCB 和达到体外细胞传代限次的细胞(Limit of In Vitro Cell Age,LIVCA))的病毒检测方法举例见表 1。
3.1.1 主细胞库
应对 MCB 进行内源性和外源性病毒污染的全面筛查。对于含一种或一种以上人或非人灵长目动物配体的异种杂交细胞系,由于这些细胞引起的病毒污染可能造成特殊的危害,应进行人或非人灵长目病毒的测定。
外源病毒的检测应包括表 1 中所述的广义和特异性病毒检测试验。鼓励引入用于检测多种外源病毒的新方法。为确保测出污染性病毒,测定方法应基于细胞系的来源和历史,以及在细胞系生成和 MCB 扩增过程中暴露于人或动物来源材料的可能性。
3.1.2 工作细胞库
应根据表 1 对每个 WCB 进行外源病毒检测。如果已对 MCB 进行相应的外源病毒检测,并且对己达到或超过 LIVCA 的 WCB 来源的细胞进行过外源病毒检测,则不必再对原有的 WCB 作类似检测。一般不建议针对 WCB 进行抗体产生试验。另外, 不检测 MCB,而对每个 WCB 进行全面检测来作为替代手段也是可以的。
3.1.3 达到体外细胞传代限次的生产用细胞
应根据在试生产规模或商业化生产规模条件下达到或超过设定的体外细胞传代限次的生产细胞的数据来确定用于生产的 LIVCA。一般通过扩增 WCB 获得生产细胞,MCB 也可直接用于制备生产细胞。有些内源病毒可能在 MCB 阶段没有被检测出,应对达到 LIVCA 的细胞的内源病毒进行评价。达到 LIVCA 的细胞也称为生产终末细胞(End of Production Cell,EOPC)。针对用于生产的达到 LIVCA 的细胞至少应进行一次适当的检测(如表 1 所示),以进一步确保生产工艺不会导致内源病毒激活或外源病毒扩增(包括生长缓慢的病毒)。如果此时测出有外源病毒污染,应对工艺流程进行仔细检查以确定污染源。
3.2 对病毒检测和鉴别方法的建议
对内源病毒和外源病毒的检测方法有很多。表 2 列举了一些检测方法。建议采用此类检测方法,但表中所列方法并非全部且并非固定不变。大部分适用的方法会随科学的发展而变化,因此在有充分资料支持的情况下可采用替代方法。鼓励制造商与相应的监管机构讨论这些可替代性方法。一个全面测定策略应包括以下考虑要点:细胞系来源;传代历史;在细胞系生成、细胞库制备以及生产过程中使用的生产用原材料和试剂。该策略还应包括根据对细胞基质、原材料和所用试剂的风险评估进行适当的其他检测。例如,如果存在特异性病毒的可能性相对较高,除非另有说明,否则应包括针对该病毒的特定检测或其他方法。应设相应的对照试验,以证明试验具有充分的敏感性和特异性。
下一代测序(Next Generation Sequencing,NGS)技术和核酸扩增技术(Nucleic Acid Amplification Techniques,NAT),如聚合酶链式反应(Polymerase Chain Reaction,PCR)可能分别适用于广谱和特定病毒检测。采用此类试验时,无需将其与当前推荐的体外和体内检测方法进行系统的头对头比较。特别是,不建议对体内检测方法进行头对头比较,以满足替代、减少、和优化动物使用的全球目标。由于检测方法的灵敏度和病毒检测的广度,NGS 也可用作基于细胞的感染性试验的替代方法,以解决潜在的检测局限性,或用于检测试验系统中无可见表型的病毒。对阳性结果应进行研究,以确定检测到的核酸是否与感染性病毒相关。
以下是针对制造商应使用的一般框架的简要描述,用以开发一个全面的针对产品和生产工艺特定(或适当)的病毒检测方案。检测计划或策略应随附检测方法的适当理由。
3.2.1 逆转录病毒检测
应对 MCB 和用于生产的达到或超过 LIVCA 的细胞进行逆转录病毒检测。这些检测包括通过直接接种或共培养进行的感染性测定、逆转录酶(Reverse Transcriptase,RT)活性测定和通过透射电子显微镜(Transmission Electron Microscopy,TEM)对颗粒进行评价。
如果不知道细胞系是否产生逆转录病毒颗粒,则应对细胞进行 TEM 检测,并对澄清的上清液进行基于 PCR 的 RT 测定(如产物增强型 RT 测定法)。基于 PCR 的 RT 测定法极具应用价值,原因在于其可以检测所有逆转录病毒的 RT 活性;然而,RT 活性可能与感染性或非感染性逆转录病毒有关。由于一些细胞 DNA 聚合酶可发生交叉反应并导致阳性 RT 结果,应在确认 RT 活性(由于逆转录病毒污染)或阳性 TEM 结果后进行试验,以在认可有效的细胞中对感染性逆转录病毒进行检测,包括采用读出灵敏测定法对人体细胞系进行逆转录病毒检测。
如果已知细胞系能结构性产生逆转录病毒颗粒(如来源于啮齿类动物、昆虫和鸟类细胞系),则预期会存在 RT 活性,因此可能不需要应用基于 PCR 的 RT 测定法。应进行 TEM 检测以检查存在的逆转录病毒颗粒的类型(如 A 型和 C 型),并使用相关的认可有效的细胞(如用于啮齿类动物逆转录病毒的杜氏小家鼠细胞和SC-1(小鼠胚胎细胞)),采用用于逆转录病毒检测的读出灵敏测定法(如产物增强型逆转录酶活性(RT)测定法,肉瘤-阳性、白血病-阴性(S+L-)测定法,或 XC 空斑试验或广义分子检测)进行感染性检测,以确定内源性逆转录病毒颗粒是否具有感染性。
判读逆转录病毒检测结果时应将所有可用数据纳入考量。基于第 3.3 节和第 5 节中讨论的风险评价,表达内源性逆转录病毒颗粒的细胞系或可应用于生产。
尚未发现转导研究在已充分表征内源性逆转录病毒(例如中国仓鼠卵巢(CHO)、NS0 和 Sp2/0)的细胞系中的应用价值。然而,此类研究可能有助于对新细胞基质是否存在未知的内源性逆转录病毒进行评价。此外,基于风险评估,对潜伏性 DNA 病毒(例如,人类细胞中的疱疹病毒)和潜伏性 RNA 病毒(例如,昆虫细胞中的诺达病毒)进行转导研究也可能是适当的。这些研究可能有助于为来源于新细胞基质的产品提供病毒检测和清除策略。
3.2.2 体外细胞培养感染性检测
进行体外检测时,应将供试品(见表 2)接种到多种敏感的细胞培养物中,以确保能较为广泛的检测到人和有关动物的病毒。检测中所用细胞应根据待测细胞基质的物种来源的风险评估而定。细胞系组应包括来源物种的细胞系以及对人类病毒敏感的人和非人灵长目动物细胞系。
感染性检测的性质和待测样本应根据细胞的来源或细胞处理情况中可能存在的病毒类型而定。对于细胞系检定,应在 14 天初始细胞培养检测后进行二次传代,持续时间为 14 天,随后观察致细胞病变病毒和红细胞吸附/红细胞凝集病毒。
或者,分子病毒检测方法可用于补充(例如,需要解决某些限制,如供试品介导的干扰或毒性)或替代细胞培养检测法。
3.2.3 体内检测
基于 NGS 进行病毒检测的广度,及其对替代、减少和优化动物试验使用的全球目标的促进作用,因此鼓励使用 NGS 替代体内检测。采用 NGS 替代体内检测时可能需要提供相关验证研究数据包以作为支撑。根据风险评估和总体检测策略,体内检测可能包括将供试品(见表 2)接种到哺乳期小鼠、成年小鼠和鸡胚中。还可根据所检测细胞系性质和来源使用其他动物物种进行试验。应对受试动物的健康情况进行监测,如有异常应进行调查以确定病因。
3.2.4 抗体产生试验
当存在暴露于特定物种的病毒的可能性时,应进行抗体产生试验。例如,通过将供试品(见表 2)接种至无特定病原体(SPF)的动物(如小鼠、大鼠,以及随后用于检测特异性药物抗体的仓鼠)中,可检测啮齿类动物源性细胞系中是否存在啮齿类动物病毒或通过啮齿类动物传代产生的病毒以及可能来源于啮齿类动物材料的试剂。这种试验包括:小鼠抗体产生(MAP)试验、大鼠抗体产生(RAP)试验和仓鼠抗体产生(HAP)试验。目前,有关用抗体产生试验筛查出的病毒见表 3。
病毒特异性 PCR 或靶向分子生物学方法可用作表 3 中所述动物试验的替代检测方法。
3.2.5 分子生物学方法
分子生物学方法可用于补充或替代基于体外细胞培养试验和体内动物试验。
3.2.5.1 核酸扩增技术
核酸扩增技术(NAT),如基于 PCR 的方法,通常单独使用或以多重形式使用,以检测已知病毒或已知密切相关病毒家族的病毒序列。靶向 NGS 方法也可能适用于已知病毒的灵敏检测。这些分子生物学方法可用于补充细胞培养检测法,其中由于检测干扰而存在局限性,并且在此类病毒无法在细胞培养物中轻易生长以进行感染性检测的情况下,其为检测特异性病毒的有效工具。NAT 方法还适用于更广谱的病毒检测(例如,简并 PCR),然而特异性可能会降低。由于检测的特异性,可能需要进行大量的病毒特异性 PCR 检测,以检测由单个普遍应用的生物测定法检测到的病毒的广度。应针对 NAT 检测方法的预期用途进行了适当的鉴定或验证。
3.2.5.2 下一代测序
新的先进分子生物学方法,如 NGS(高通量测序),已被证明有能力进行广谱的病毒检测。NGS 可以提供确定的病毒检测的灵敏度和广度,并减少动物的使用和缩短检测时间。应提供所用 NGS 方法的验证材料以支持 NGS 方法在申请中使用。其包括方法验证以及检测方法或基质特异性鉴定(如适用)。基于潜在的安全问题,生物信息学分析可以针对特异性病毒或未知病毒进行广谱的病毒检测。NGS 可替代体内试验进行广谱的病毒检测,用于检测未知或非预期的病毒种类。NGS 也可以补充或替代体外细胞培养试验,用于检测已知、未知或非预期的病毒种类。此外,该检测法还可用于检测已知病毒,并替代 HAP、MAP 和 RAP 试验以及其他病毒特异性 PCR 检测法。
对于细胞基质和细胞库的表征或检测、已知和未知病毒的检测,以及由于载体病毒(见附件 7)缺乏有效中和或由于产品或各培养基组分引起的毒性而存在测定干扰的病毒种子或收获物的情况下,应考虑使用 NGS。在这类应用中,NGS 可用于检测存在于细胞 DNA 中的病毒序列(基因组学)或在细胞中以 RNA 表达的病毒序列(转录组学),以及用于检测存在于病毒颗粒中的病毒基因组(病毒学)。应提供选择这些不同策略的依据。
应用 NGS 对已知病毒进行灵敏检测和/或对新型病毒进行广谱检测时,申请人应考虑 NGS 工作流程中的几个关键步骤。这些步骤包括:1)基于样本材料类型的样本处理(检测时)和加工;2)高效的病毒核酸提取(包括包膜和非包膜颗粒)和文库制备;3)选择合适的测序平台,以及 4)针对多样性数据库(具有不同病毒家族的病毒序列)进行全面的生物信息学分析。样本处理和加工期间执行的所有步骤的目的在于最大限度地进行病毒检测。
应使用合适的标准品或参照品进行分析鉴定和验证,以评估方法中所涉及不同步骤的性能,并证明病毒检测的灵敏度、特异性和广度。这可能包括使用目前可用的具有不同物理(大小、有包膜和无包膜)、化学抗性(低、中和高)和基因组(DNA、RNA,双链和单链、线性、环状)特征的病毒试剂标准品,以评估整个 NGS 工作流程或特定步骤的性能,应使用具有不同病毒序列的综合病毒数据库进行广谱的病毒检测。此外,其他标准品类型可用于评估特定技术和生物信息学步骤。由于 NGS 的工作流程复杂,我们鼓励制造商与相应的监管机构就方法验证和数据提交相关期望进行讨论。
3.3 细胞系的可接受性
用于生产的某些细胞系含有内源性逆转录病毒及其他病毒或病毒序列,这些病毒可能被重新激活为感染性病毒。为此,第 5 节中介绍了建议的生产行动计划。对于含有内源性逆转录病毒以外的病毒的细胞系的可接受性,应由相应的监管机构根据产品的收益、产品的预期临床用途、污染病毒的性质、对人类潜在的感染性和致病性、产品的纯化工艺(如病毒清除评价资料)及对纯化后产品进行病毒检测的程度,逐一进行风险受益分析后综合考虑。
4. 未加工收获液的病毒检测
建议制造商制定计划,持续评估生产批次中的外源病毒。未加工收获液病毒检测的范围和程度应结合以下几点综合考虑后作出决定:用于生产所需产品的细胞系性质,细胞系检定过程中所执行病毒检测的结果和程度,培养方法,原材料和试剂来源以及病毒清除研究结果等。
未加工收获液包括集中回收的一批或多批细胞和培养基。在进行深加工前,从生产反应器中获取的具有代表性的未加工样本,最适合于检查外源病毒污染,若存在病毒污染,在该步骤最有可能被检测出来。应对未加工收获液进行适当的病毒检测。对于灌注或连续生产工艺,细胞可能不易获取(例如,因使用中空纤维或类似的微滤系统)。在这种情况下,未加工收获液是从生物反应器中收获的液体。应考虑细胞分离技术和渐进式过滤器污染对这些未加工收获液的代表性的潜在影响。如果未加工收获液在供试细胞培养物中具有毒性,则可以考虑部分初加工,例如最小样本稀释或替代试验(见第 3.2 节)。在某些情况下,可对同时含有完整细胞和破碎细胞以及深加工前从反应器中取出的细胞培养物上清液组成的混合物进行测定。对于涉及连续收获的工艺,应证明取样策略(包括样本的周期性和组成)的合理性,原因在于外源病毒和内源病毒颗粒有可能随着细胞培养持续时间的不同而发生变化(见第 7 节)。
应对各未加工收获液进行常规外源病毒检测。这可能包括使用数种细胞系进行的体外筛选试验或广谱的分子病毒检测方法(如 NGS)(见第 3.2 节)。基于风险评估(考虑到细胞基质、动物源性原材料或试剂的使用以及工艺的病毒清除水平),应对指示细胞培养物进行至少两周的观察。根据风险评估,对特异性病毒或病毒家族的检测也可能适当包括在内(如小鼠小病毒)。在适当情况下,也可选择 PCR 或其他分子生物学方法,快速试验方法可以促进实时决策。
如果在未加工收获液中检测到任何外源病毒,除非提供合理证明,否则不得将收获物用于产品生产(关于在收获物中检测到外源病毒的材料使用指南,参见第 5 节。)。应仔细检查工艺,以确定污染的根本原因和污染程度,且应采取适当的措施。对于连续生产工艺,最终亚批的放行要求证明收获培养物液体用于生产该亚批期间不存在病毒污染。如果检测到外源病毒,应考虑对潜在污染材料进行隔离,以降低更广泛的生产影响。
5. 对纯化后产品进行病毒清除研究和病毒检测的依据和行动计划
病毒检测方案应覆盖 MCB、药品生产的各个阶段直到最终产品产生的整个过程,设计的方案应具有相关性和合理性,包括对未加工收获液病毒清除的评价和表征。病毒清除的评价和表征在病毒检测方案中起着关键作用。其目标应是获得最佳合理的保证,即确保产品不受病毒污染。
在选择清除研究用病毒时,首先要明确对己知病毒清除的工艺评价和对用非特异“模型”病毒(后文叙述)进行的工艺病毒清除验证的评估之间的区别。关于相关模型病毒、特异模型病毒和非特异“模型”病毒的定义,见术语解释。工艺评价需了解在该工艺过程(如未加工收获液)中可能存在的病毒量,以及清除多少量才能确保产品的安全性。了解灭活过程的时间依赖性将有助于确保灭活工艺的有效性。在对己知污染物的清除能力进行评价时,需对灭活的时间依赖性进行深入研究,对灭活或清除方法的重现性加以证实,同时还应对工艺参数作出评价。当使用非特异“模型”病毒对生产工艺清除病毒的稳健性进行表征时,研究设计中应对非包膜病毒予以特别的关注。表征研究中病毒清除的程度可能受到细胞系和未加工收获液检测结果的影响。这些研究应按以下所述方法进行(见第 6 节)。
表 4 提供了针对细胞或未加工收获液病毒检测结果而采取的行动计划示例。该计划包括工艺评价和病毒清除的鉴定以及对纯化后产品进行病毒检测。表中列出了各种情况,并在下文加以说明。其中应使用非特异“模型”病毒对病毒清除情况进行鉴定。A、B 两种情况是最常见的。被病毒污染的生产系统通常不能用于生产,啮齿类动物逆转录病毒污染除外。如果要将 C、D、E 三种情况的细胞系用于药物生产,则需有充分正当理由,并须与适当的监管机构进行讨论。当使用 C、D、E 情况中细胞系时,须有己经过验证且有效的步骤,确保所疑病毒从生产工艺中灭活或去除。
情况 A: 当细胞或未加工收获液中未发现有病毒、病毒样颗粒或逆转录病毒样颗粒时,应使用上述非特异“模型”病毒进行病毒去除和病毒灭活研究。
情况 B: 在啮齿类动物细胞系中,如果仅存在啮齿类动物逆转录病毒(或非致病性逆转录病毒样颗粒,如啮齿 A 型和 R 型颗粒),应使用特异“模型”病毒(如鼠白血病病毒)进行工艺评价。对于纯化后产品,应使用具有高特异性和高敏感性的合适方法对所疑病毒进行检测。申请上市时,应至少提供三批试生产规模或生产规模的纯化后产品的检定数据。对于常用作药物生产基质的中国仓鼠卵巢(CHO)、C127、BHK 等细胞系和鼠杂交瘤细胞系, 尚未报告与产品病毒污染相关的安全问题。对于这些细胞系,由于其内源性颗粒己进行广泛表征,病毒清除问题也己证实,通常不建议再检测纯化后产品或原料药中的非感染性颗粒。使用如情况 A 中所述的非特异“模型”病毒进行研究即可。类似的方法可能适用于产生内源性逆转录病毒样颗粒(已进行广泛表征)的昆虫细胞系(例如 Sf9)。
情况 C: 已知细胞或未加工收获液中含有除啮齿类动物逆转录病毒以外的病毒,而这些病毒又无证据证明其对人有感染性(如Sf9 弹状病毒(如表 3 脚注 2 中所标明,除啮齿类动物逆转录病毒(情况 B)以外的病毒)),则病毒去除和灭活的评价研究应使用已鉴定的病毒。如果不能使用己鉴定的病毒,应使用“相关”病毒或特异“模型”病毒来评估其工艺清除效果是否可被接受。在灭活的关键步骤,应对己鉴定的病毒(或“相关”病毒或特异“模型”病毒)进行时间依赖性地灭活,作为对这些病毒工艺评价的一部分。对于纯化后产品,应使用具有高特异性和高敏感性的合适方法对所疑病毒进行检测。申请上市时,应至少提供三批试生产规模或生产规模纯化后产品的检定数据。
情况 D: 当检测出已知病毒对人类具有感染性(例如表 3 脚注1 所指示的病毒)时,则该产品应仅在特殊情况下才可接受。在这种情况下,应使用己鉴定的病毒进行病毒去除和灭活评价研究,并使用具有高特异性和高敏感性的特殊方法对所疑病毒进行检测。如果不能使用己鉴定的病毒,应使用相关和/或特异“模型”病毒(后文叙述)。应证明在工艺纯化和病毒灭活过程中,己去除和灭活所选病毒。应在灭活的关键步骤取得时效性灭活数据,作为工艺评价的一部分。对于纯化后产品,应使用具有高特异性和高敏感性的合适方法对所疑病毒进行检测。申请上市时,应至少提供三批试生产规模或生产规模的纯化后产品的检定数据。
情况 E: 在细胞或未加工收获液中检测到用现有方法无法分类的病毒时,该产品一般不予接受,因为这种病毒可能具有致病性。在极个别的情况下,并有充分正当理由说明该细胞系能用于药物生产时,在进入下一步之前,须与监管机构进行讨论。
情况 F: 当辅助病毒用于生产时,应使用辅助病毒本身或特异模型病毒(例如杆状病毒、腺病毒或疱疹病毒)证明病毒清除情况。
6. 病毒清除工艺的评价和鉴定
病毒清除或灭活工艺的评价和鉴定对生物技术产品的安全性起着重要的作用。过去发生的多例污染事件是由于使用未知或甚至未被怀疑的受污染的原材料。尽管这种情况并不发生在经充分表征的细胞系制备的产品,而是发生在从其它原材料中制备的生物制品中,,但是病毒清除评价可使我们确信那些未知的、预想不到和有害的病毒都有可能被清除。研究必须被详细记录并在受控条件下进行。
病毒清除研究的目的是:1)评估有效灭活或去除病毒的工艺步骤,以及 2)定量估计通过该工艺获得的病毒的整体降低水平。具体做法是有目的地将一定量的病毒加入(即“加标”)到原料或各工艺步骤的抽样样本中,通过研究在后续工艺步骤样本中的病毒量来验证该工艺步骤对病毒去除或灭活的效果。如果使用较少工艺步骤就能证明其可充分清除病毒,则无需对生产工艺的每一步进行评价和鉴定。应注意的是,生产的其他工艺步骤可能会对己取得的病毒灭活或去除结果产生间接影响。制造商应解释并证明研究中用于评价病毒清除的方法的合理性。一般而言,为了确定进入纯化工艺的内源病毒颗粒的数量,应对三个批量或批次的细胞培养活动进行定量。这些数据应作为上市申请或注册资料的一部分提交。
降低病毒感染性可以通过清除病毒颗粒或灭活病毒的感染性的方式达到。对于所评估每一生产工艺步骤,应说明病毒感染性丧失的可能机制,即是被灭活还是被去除。对于灭活步骤,应对研究进行规划,在不同时间取样并建立灭活曲线(见第 6.2.5 节)。
病毒清除评价研究的目的是:1)证明 MCB 中己知存在的病毒已清除,或 2)确保无法检出或可能进入生产过程的外源病毒已清除。下降因子一般以对数标度表示,说明虽然病毒的残留感染性永远也不可能降至 0,但对数级可以大幅下降。
除了对已知存在的病毒进行清除研究外,还应进行研究以确定去除或灭活其他病毒的能力。使用具有一系列未知或非预期生化和生物物理特性的病毒进行研究的目的是确定生产工艺的稳健性,而不是实现特定的灭活或去除目标。应对生产工艺步骤中的病毒灭活或去除的能力加以证实(见第 6.3 节)。此类研究的目的并非对特定安全风险进行评价。因此,无须达到特定的清除值。
6.1 用于评价和鉴定病毒清除率的病毒选择
为了测试生产工艺清除病毒的总体能力,供清除评价和工艺鉴定研究用的病毒应与可能污染产品的病毒相似,而且要有广泛的理化特性。制造商应根据本指导原则中提供的评价和鉴定研究的目的对病毒的选择作出合理解释。
6.1.1 “相关”病毒和“模型”病毒
病毒清除研究中的一个主要问题是确定使用何种病毒。这些病毒可分为三类:1)“相关”病毒,2)特异“模型”病毒和 3)非特异“模型”病毒。
“相关”病毒是指用于生产过程中评价病毒清除情况的病毒,可以是已被鉴定的病毒,或是与已知病毒种类相同的病毒,或是可能会污染细胞培养物或污染生产过程中使用的其他试剂或材料的病毒。应证明纯化和/或灭活工艺能去除和/或灭活此种病毒。如果得不到“相关”病毒,或不太适用于病毒清除的工艺评价研究(如不能离体培养到足够的滴度),应使用特异“模型”病毒代替。适用的特异“模型”病毒是与己知病毒或可疑病毒密切相关(同种或同属),并与所观察到的或可疑的病毒具有类似理化特性的病毒。
啮齿类动物细胞系一般都含内源性逆转录病毒颗粒或逆转录病毒样颗粒,可能具有感染性(C 型颗粒),也可能没有感染性(细胞浆 A 型和 R 型颗粒)。应确定所用生产工艺具有去除和/或灭活啮齿类动物逆转录病毒的能力。此项工作可使用鼠白血病病毒作为鼠源细胞的特异“模型”病毒。
对于 CHO 细胞衍生的产品,也可使用 CHO 衍生的内源病毒颗粒进行病毒清除实验。未对这些病毒颗粒进行感染性检测,检测方法(例如,分子或生化)应经过鉴定方可使用。当通过 EB 病毒(EBV)使 B 淋巴细胞永生化而获得分泌单克隆抗体的人细胞系时,应确定生产工艺去除和/或灭活疱疹病毒的能力。伪狂犬病毒也可用作特异“模型”病毒。
当研究目的是确定生产工艺去除和/或灭活病毒的总体能力(即确定病毒清除工艺的稳健性)时,应使用具有不同特性的非特异“模型”病毒进行病毒清除鉴定研究。从“相关”病毒和/或特异“模型”病毒研究中所获取的研究数据也有助于这种评估。一般不需要对所有病毒进行测试,应对那些对物理和/或化学方法处理具有显著耐受性的病毒加以关注,因为从这些病毒所获取的结果可为评价生产工艺去除和/或灭活病毒的总体能力提供有用的信息。选用病毒的种类和数量与细胞系质量和特性及生产工艺有关。
附件 2 和表 A-1 例举了代表不同理化结构的“模型”病毒以及己用于病毒清除研究的一些病毒。
6.1.2 其他考虑
其他需要考虑的问题如下:
- 尽管有时不可能,能培养出高滴度的病毒是最理想的;
- 应有一种有效和可靠的测定方法对要测试的每一道生产工艺中所用的每种病毒进行检测;有些病毒可能会对从事病毒清除研究的人员造成健康损害,对此应加以重视。
6.2 病毒清除评价和鉴定研究的设计和内涵
6.2.1 设施和人员
根据药品生产质量管理规范规定,不能将任何病毒引入生产设施。因此病毒消除研究应在隔离的实验室进行。该实验室应进行病毒学研究并由具有病毒学专业知识的人员会同参与设计和制备缩小规模的纯化工艺的生产人员一起进行。
6.2.2 缩小规模的生产体系
应对缩小生产规模的有效性加以证实。缩小规模的纯化工艺水平应尽量接近实际生产水平。色谱设备、柱床高度、线性流速、流速/柱床体积比(即过柱时间)、缓冲液、凝胶类型、pH、温度、蛋白浓度、盐及产品均应代表生产规模水平。应有一个类似的洗脱方案。对于其他生产工艺步骤,亦应有类似考虑。有些偏差是不可避免的,但应重视其对结果的影响。
6.2.3 对病毒逐步消除的分析
在进行病毒清除研究时,应考虑评估一个以上生产工艺步骤对病毒消除的综合作用,还应对每个可能参与病毒清除的生产工艺步骤的去除和灭活病毒的能力进行分别评估,并考虑各个工艺步骤的确切作用。每一工艺步骤的测试样本中应含足够量的病毒,以便对每一工艺步骤的有效性进行充分评估。一般来说,应将病毒加入到每一步生产工艺中的待测样本中。在某些情况下,需将高滴度病毒加入未加工收获液中,并在随后各步骤测定病毒浓度即可。当通过分离步骤完成病毒去除时,建议在适当和可能的情况下,研究病毒载量在不同分离部分中的分布情况。当在生产过程中有多个生产工艺步骤使用杀病毒缓冲液时,也可用替代的方法,如用较弱的杀病毒缓冲液进行平行加标,作为对生产工艺总体评估的研究组成部分。另外应对每一被评估工艺步骤之前和之后的病毒滴度进行测定。感染性定量测定方法应有足够的敏感性和重现性,还要有足够的重复实验以确保结果具有充分的统计学可信度。如有正当理由,也可使用与感染性无关的定量分析。在进行感染性测定时,要有相应的病毒对照组以确保方法的敏感性。如抽样病毒的浓度较低,其统计结果也应该考虑(见附件 3)。
6.2.4 确定物理去除或灭活
可通过去除病毒或灭活病毒降低病毒感染性。对于所评估的每一生产工艺步骤,应说明病毒感染性丧失的可能机制,即灭活还是被去除。如所用生产工艺清除病毒感染性的能力较低,而病毒清除对于产品安全又是一个重要因素,则应针对性或额外地引入灭活/去除步骤。对某一特定步骤,区分其是去除还是灭活作用可能是必要的。例如,当多个病毒清除步骤中使用的某种缓冲液可能有灭活作用(即,多个色谱步骤共同使用的缓冲液有灭活作用)时,应明确每个色谱步骤对病毒的去除效果。
6.2.5 灭活评估
为了对病毒灭活作出评估,应在未加工的原料或中间品中加标感染性病毒,并计算下降因子。要知道病毒灭活不是一个简单的一级反应,通常比较复杂,包含有快的“一期”反应和慢的“二期”反应。因此,要在不同的时间点取样研究并建立灭活曲线。建议灭活研究中,取样点除最低作用时间外,至少还应设一个大于零和小于最低作用时间的时间点。如果该病毒是一种己知人致病性“相关”病毒,研究工作必须取得更多的数据,并设计出一种有效的灭活工艺。但是,如使用非特异“模型”病毒进行灭活研究,或使用特异“模型”病毒作为病毒颗粒的替代物,如 CHO 细胞浆内逆转录病毒样颗粒,必须至少分别作两次独立的研究来证实清除工艺的可重复性。如可能,应根据可在加标起始物料中检测到的病毒量来确定初始病毒载量。如果不可行,可根据加入病毒的滴度计算初始病毒载量。如果由于灭活太快,来不及根据工艺条件建立灭活曲线,应进行相应的对照试验以证实病毒经灭活处理己失去感染性。
6.2.6 分离柱的功能和再生
纯化方案中的色谱分离柱和其他设备经一段时间反复使用后,其清除病毒的能力会发生变化。应指明色谱介质/树脂的使用周期,并确定影响病毒清除的关键工艺参数。
对于蛋白 A 亲和捕获色谱,先验知识表明,对于使用过的(例如,寿命结束)色谱介质/树脂,其病毒去除效率不受影响或略有增加。因此,不期望使用已使用过的树脂进行产品特定研究。先验知识也可能适用于病毒清除中涉及的其它色谱类型(例如,阴离子交换或阳离子交换)。因此,为支持树脂可重复用于其它色谱类型,应当提供包括内部经验在内的等效先验知识以及详细的依据,而不是用使用寿命结束的树脂开展产品特定病毒清除研究。
在设备再次使用前,必须保证生产设备可能残留的任何病毒都被充分消灭或去除。例如,可提供证据证明色谱柱经过清洗和再生过程可灭活或去除病毒。
6.2.7 特别注意事项
应考虑下述特别注意事项:
- 制备高滴度病毒时应注意避免凝集反应,凝集的病毒更容易被物理方式去除,但同时更难被灭活,因此,与实际生产不符;
- 须注意有效分析方法的最低检出病毒量;
- 应进行平行对照分析研究,以评估样本是否在检测病毒滴度前因稀释、浓缩、过滤或贮存等原因使病毒失去感染性;
- 加入产品中的病毒“加标”体积要小,从而不会使产品稀释或改变产品的性质。经稀释后的试验蛋白样本己不再与生产规模的产品相同;
- 诸如缓冲液、培养基或试剂等的微小差异都会对病毒清除产生重大影响;
- 病毒灭活具有时间依赖性,因此,加标产品在特定缓冲溶液中或特定色谱分离柱中停留的时间长短应能反映生产规模的工艺条件;
- 应分别对缓冲溶液和产品评估其毒性或对病毒滴度检测方法的干扰,因为这些因素可能会对指示细胞产生不良影响。如果溶液对指示细胞有毒性,可能需要进行稀释、调整 pH 值或者将含有加标病毒的缓冲液进行透析。如果产品本身具有抗病毒活性,进行病毒清除研究时,应采用无产品的空白对照试验,即使不用产品或用无抗病毒活性的相似蛋白替代品会影响某些生产工序的病毒行为。应纳入足够的对照品,以证明如透析、贮存等准备检测样本的单个过程对去除/灭活加标病毒的影响;
- 许多纯化方案中都反复使用相同或相似的缓冲液或分离柱。分析数据时,应考虑这种纯化方法所带来的影响。某一特定工艺的病毒消除有效性会随所在的生产阶段而有所不同;以及
- 当生产条件或缓冲剂具有很强细胞毒性或杀病毒作用时,可能会低估总体下降因子,因此需逐例讨论。由于病毒清除研究本身的局限性或因设计不够完善,也会高估总体下降因子。
6.3 病毒清除研究的说明
评估病毒灭活/去除的目的是对各工艺步骤作出评价和鉴定,了解其对灭活/去除病毒是否有效,并对生产过程中病毒下降的总体水平进行定量分析。就情况 B-E 所述的病毒污染而言,不仅要证明病毒己被消除或灭活,而且还要证明纯化工艺具有足够能力将病毒清除,从而确保最终产品具有相当的安全性。生产过程中所消除或灭活的病毒量应与未加工收获液中可能存在的病毒量相比较。
为了进行这种比较,必须对未加工收获液中的病毒总量作出估计。可采用检测感染性或其他诸如 TEM 或 NAT 的方法进行估计。整个纯化工艺应能够清除比未加工收获液的同等单剂量中的估计量更多的病毒。病毒下降因子计算见附件 4;每个剂量颗粒的估算见附件 5。制造商应认识到,不同种类的病毒,其清除机制有可能不同。当判断病毒灭活/去除过程的有效性时,应综合考虑以下各种因素:包括:
- 所用测试病毒是否合适;
- 消除研究的设计;
- 所获得的 log 下降值;
- 灭活时间对于病毒灭活效果的影响
- 工艺参数变化对病毒灭活/去除的潜在影响;
- 测定方法灵敏度范围;灭活/去除方法对某些种类病毒可能具有的选择性
建议设计一种能清除各种潜在病毒污染物的下游工艺。在这种情况下,只要可行且不会对产品产生不利影响,建议实施两个不同的有效步骤,在其作用方式上相互补充。其中一个生产步骤应能有效清除无包膜病毒。一个有效的病毒清除步骤通常会使病毒量可重复地减少,至少在两个独立研究中显示为 4 对数或更多。然而,认识到可重复降低病毒载量(数量级为 1-3 log)的步骤有助于确保病毒安全性,可考虑用于评估总体病毒降低情况。专门用于病毒灭活/去除的工艺步骤(例如溶剂/洗涤剂处理、仅用洗涤剂处理、病毒过滤(纳米过滤)或在低 pH 值下培养),在清除各种病毒方面非常有效。使用为去除小病毒而设计的病毒过滤器,对较小的细小病毒或多瘤病毒来说也是一个强有力的病毒清除步骤。最后,有经验表明,在纯化单克隆抗体的蛋白 A 捕获步骤之后,通过在低 pH 值下培养,可以有效灭活异嗜性鼠白血病病毒(XMuLV)和伪狂犬病毒。
以下任何一种方法都能实现可接受的总体清除率:多步灭活法,多步互补分离法,或灭活与分离结合法。由于分离法对病毒的物理化学特性具有很强的依赖性,这影响病毒与色谱固定相(如树脂或色谱膜)的相互作用和沉淀特性,因此“模型”病毒可用与靶病毒不同的方法进行分离。应正确定义和控制影响分离的生产参数。表面特性的变化,如糖基化,也是差异的来源。但是尽管存在这些潜在的可变因素,仍可采用将几种互补分离过程结合的方法或将灭活与分离相结合的方法达到有效清除病毒目的。因此,色谱层析分离、过滤和提取等方法,只要控制条件并完善设计,都是有效清除病毒的方法。
总体下降因子一般以各因子之和来表示。除另有合理解释,病毒滴度下降在 1 log10 或以下可忽略不计。
如所用生产工艺去除病毒感染性的能力较低,而病毒清除对于产品安全又是一个重要因素,则应另外增加一次或多次有针对性的灭活/去除步骤。制造商应对所有病毒下降因子的合理性作出说明,并将根据以上所列因子对结果作出评价。在评估结果时将考虑上述因素。
6.4 病毒清除研究的局限性
病毒清除研究有助于保证最终产品达到可接受的安全水平,但其本身并不能完全安全可靠。这是由于病毒清除研究设计和执行过程中的许多因素会使对生产工艺清除病毒感染的能力作出不正确的估计,其中包括如下因素:
- 用于某一生产工艺病毒清除研究的病毒制品通常来自特定的细胞培养物。这种病毒在生产步骤中的行为可能与来自细胞培养基中生物原材料的天然病毒污染物或在生产细胞中复制的病毒污染物的行为不同。例如,天然病毒和培养病毒在纯度或凝集度上是不同的;
- 病毒感染性的灭活常是一条双相曲线:先有一个快速起始阶段,接着是一个较慢的阶段。这就可能使最初灭活阶段逃逸的病毒对以后各阶段具有更强的耐受性。如具有耐受性的那一部分病毒以病毒凝集物的形式出现,则其感染性可能对许多不同的化学处理及热处理都产生耐受性;
- 生产工艺清除或灭活病毒的总体清除能力以每一工艺步骤病毒下降 log 值之和表示。将各步骤的下降因子相加,尤其是将下降较少(如低于 1 log10)阶段的因子相加,可能会对去除病毒的实际能力估计过高。生产过程中其他有相似灭活机制的病毒下降因子的相加也可能会对整体清除病毒能力产生过高估计。此外,除非有合理的理由,否则亦不能将重复相同或几乎相同的方法所获得的下降值包含在其中;
- 下降因子用 log 滴度来表示,说明尽管残留的病毒感染性可能己大大降低,但是决不会降至 0。例如,当每毫升含 8 log10 感染单位的制品感染性下降 8 log10,其结果是每毫升 0 log10 或是每毫升一个感染单位,此时应考虑测定方法的检测范围是否符合要;
- 尽管设计缩小生产规模时慎之又慎,但试生产规模工艺还是可能与商业化生产规模工艺有所不同。
6.5 统计
病毒清除研究结果评估时应引入统计学分析。研究结果应具有统计学意义才能支持所得出的结论(见附件 3)。
6.6 应用先验知识评价病毒清除情况
一般而言,病毒清除情况通过实验来评价,即把病毒添加到待研究的每个步骤下特定产品在加工样本中。如果制造商正在通过既定且鉴定良好的工艺(即使用相同平台技术)开发类似产品,则其他产品生成的病毒清除数据可能适用于同一工艺步骤的新产品。然而,为了利用该工艺步骤的数据,必须充分了解该工艺步骤。应明确论证特定工艺步骤先验知识的代表性。由外部和内部经验组成的先验知识应涵盖下述方面:
- 应该了解病毒清除的基本机制;
- 应该了解可能影响病毒清除的所有工艺参数;
- 应该明确病毒和产品之间的相互作用不会影响病毒清除。
- 特定工艺中间体的组成可能会影响病毒清除。对于某些工艺步骤,即使在诸如缓冲液、培养基、试剂、杂质水平和图谱等方面存在微小差异,也可能对病毒清除产生重大影响。因此,其他产品的工艺中间体组成的代表性应得以证明。此外,除非先验知识表明关于工艺中间体组成与病毒清除具有稳健性,否则新产品和已确立产品特定步骤之前的加工应遵循相似的策略;当将先验知识应用于特定产品时,应考虑上述第 6.4 节中概述的病毒清除研究的一般局限性。
外部先验知识(包括已公布数据)可用于支持灭活/去除病毒步骤的能力,并可以提供有关机制的见解。此类数据也可用于确定关键工艺参数,并为特异性病毒清除步骤中的测试设定最坏情况限值。在最坏情况下进行病毒清除研究有助于减少特定产品实验的数量。然而,将已公布的下降因子应用于特定产品时,需要通过证明所涉及不同产品生产过程具有可比性、产品中间体具有可比性予以支持,并保证产品特定属性不会影响病毒的降低水平。因此,需要仔细评估已公布数据,并且以特定平台技术的内部经验(内部先验知识)作为补充。
对于未提供采用特定产品开展病毒清除验证研究的情况下,应在具体案例具体分析的基础上,考虑药品整体病毒安全性,包括细胞基质和原材料的性质和特性,以及整体病毒清除策略,以确定其可接受性。如果数据资料不足以支持先验知识的使用,则应进行特定产品的病毒清除研究。
当使用先验知识推导 LRV 声明时,应考虑到相关平台数据中的所有 LRV,以证明此类声明的合理性。建议使用保守的 LRV 声明,以避免高估工艺步骤降低病毒能力的风险。
附件 6 提供了一些情况,即根据当前理解,来自其他产品的病毒降低水平数据的先验知识(包括内部经验)可用于说明来自相同生产平台的新产品的下降因子情况。
6.7 病毒清除的再评价
当生产或纯化工艺发生重大变化时,要考虑这种变化对病毒清除直接和间接的影响,必要时应对该工艺进行再评价。例如,生产工艺的变化可能会使细胞系产生的病毒数量发生明显变化;生产步骤的变化也可能会改变病毒清除的程度。
利用内部知识和平台概念可评估生命周期管理期间可能影响病毒清除效率的生产工艺变更。如果其他产品的内部经验不能用于特定产品和/或平台概念也不能再次应用,则必须执行特定产品的病毒清除研究。
7. 连续生产工艺的考虑要点
连续生产(Continuous Manufacturing,CM)工艺是由集成的多元的单元操作组成的动态系统,其中原材料、工艺中间体和起始物料连续进入生产工艺,产品在整个生产过程中排出。CM 可应用于部分或全部单元操作。除每个单元操作外,了解整个工艺及其动力学对于识别和降低病毒安全性相关风险至关重要。用于生产治疗性蛋白的 CM 工艺类型描述见 ICH Q13(附件 3)。
在病毒安全性方面,CM 的技术方面可能与批工艺中使用的有所不同,包括病毒的检测和去除概念、材料可追溯性、系统动力学、监测频率启动/关闭、先进工艺控制、工艺验证、工艺模型和连续工艺验证。
然而,基于工艺理解的基本原则和预期(例如基于科学和风险的方法及其控制病毒风险的实施)则与批生产相同。同时还包括污染预防策略(见第 2.2 节)。例如,当确保了与病毒清除相关的工艺参数的目标控制状态时,即使在动态工艺中灭活或去除病毒的物理和化学条件(源自批生产经验或先验知识)也适用(见第 6.6 节)。
7.1 CM 工艺中的病毒安全性
CM 工艺中病毒的控制应基于潜在污染源的风险评估(例如,起始物料和原材料以及延长的细胞培养持续时间)、工艺去除病毒的能力以及确保不存在病毒的检测能力。第 3 节和第 4 节中提供的关于检测的指南也适用于 CM。基于该评估,应制定一个策略,包括所开展的外源病毒检测的类型和频率,以证明该工艺在细胞培养和其他下游步骤是无污染的。
7.2 CM 中病毒清除的一般考虑
为了设计生产工艺和病毒清除研究,应考虑以下内容:
- 生产工艺可部分以连续或连接的操作模式运行,并且如果适用,可以使用基于批工艺的病毒清除研究设计的知识/经验来评价单元操作;
- 应评估每个单元操作和设备之间连接的潜在风险(例如,在单元操作之间使用缓冲罐或混合罐,以缓解质量流速差异或输入物料的不均匀性),以涵盖对病毒降低能力所产生的任何影响;
- 应有适当的工艺监测和取样策略来检测意外干扰或外源病毒污染。如果执行实时决策,应包括一个确定干扰或污染对输出材料质量和产品影响的程序。根据所产生的影响,应将潜在不合格物料从产品流中的转移或所生产物料的处置纳入考量;如适用,病毒清除研究-设计应考虑以下因素的潜在影响:
- o 输入物料属性的波动(例如,病毒载量、蛋白质或杂质的浓度和均一性以及聚集水平);
- o 流速、时间干扰或暂停;
- o 操作负载能力;
- o 多柱循环。
CM 还提供了针对病毒安全性需要考虑的独特方面。
7.2.1 与较长的细胞培养生产时间相关的潜在风险
在生产培养中,内源性逆转录病毒的水平可能随时间发生波动,因此应对适当的取样点进行评估,以免影响制剂的剂量风险因子计算(见第 4 节和第 3 节中有关细胞系鉴定的考虑)。
7.2.2 病毒清除研究方法
尽管预计 CM 将维持控制状态,但生产工艺将包括在启动、终止和临时工艺干扰期间工艺输出可能发生变化的时期(例如,在病毒污染的情况下,短时间内可能存在高病毒载量)。此类时间段的风险可以使用本指导原则其他部分阐述的清除研究的最佳实践来解决。针对 CM 的考虑因素包括:
- 色谱法
- o 对于重复亚批的工艺(例如,多色谱柱),一个具有合理的目标工艺条件(例如,流速、树脂负荷与色谱柱过载、树脂可清洁性)的过程可以作为缩小规模的模型;
- o 仅在所有单元操作均接受病毒清除验证的情况下,才可根据设备设计和系统集成(例如,阳离子交换层析(CIEX)的结合和洗脱模式以及阴离子交换层析(AEX)的流穿模式)选择同时验证两个或多个连接单元操作。对于连接的单元操作,如果挑战物料的载量与批操作无差异,则可以使用常规缩小规模的模型进行评价;
- 低pH值 /溶剂洗涤剂灭活
- o 在经过充分论证的目标工艺条件下进行的批工艺验证可能是合适的;
- o 对于病毒灭活(例如,pH 和溶剂/洗涤剂),应确保相关动态工艺参数的控制(例如,pH、溶剂/洗涤剂浓度、均一性和混合、温度、停留时间);
- o 当在动态过程中应用缩小规模的模型进行灭活时,应注意评价/证明规模效应(例如,停留时间分布);
- 病毒过滤
- o 如果影响病毒清除的参数设置变化不超过病毒清除研究中检测的范围(例如,最坏情况设定值),则进行的批工艺验证可能是合适的;
- o 应定义工艺控制,以允许过滤器变更和进行使用后完整性检测,同时保持病毒清除能力。应包括不中断连续工艺,以及允许在过滤器故障的情况下的物料转移。
8. 总结
本指导原则就评估病毒污染风险及产品的病毒去除方法提出建议,从而有助于从动物或人细胞系生产出安全的生物技术产品。同时强调了以下一些措施的价值:
- 对细胞基质起始物料进行全面鉴定/筛查,以明确存在哪些病毒污染物(如有);
- 通过确定人体细胞嗜性或人体感染知识评估潜在风险;
- 制定一项适当的检测未加工收获液中外源病毒的计划;
- 为获得最佳病毒清除效果,对病毒清除研究应进行仔细设计,在同一生产过程中使用不同的病毒灭活或去除方法;
- 评价病毒灭活和去除研究结果。
9. 术语表
外源性病毒
见病毒。
细胞基质
用于生产产品的细胞。
生产终末细胞(EOPC)
从 MCB 或 WCB 培养至传代水平或群体倍增水平(相当于或超过生产中达到的最高水平)收获的细胞(在与生产中使用的条件相当的条件下)。生产终末细胞为达到或超过 LIVCA 的细胞。
内源性病毒
见病毒。
体外细胞胞龄
从 MCB 复苏到生产反应器收获之间的培养时间,即以固定的稀释传代程序,得到预期的细胞群体倍增水平或者细胞传代水平所需的时间。
灭活
用化学或物理的方法使病毒感染性降低。
主细胞库(MCB)
一个细胞池的混合物,通常是在规定的条件下从选定的细胞克隆中制备的,分配到多个容器中并在规定的条件下存储。MCB 用于制备所有工作的细胞库。
主病毒种子(MVS)
主病毒种子(批或库)是疫苗病毒、辅助病毒或病毒载体,所有未来产物均将从中获得。
最低作用时间
一个处理步骤所保持的最短时间。
下一代测序(NGS)
也称为高通量测序(High Throughput Sequencing,HTS)或大规模平行测序(Massive Parallel Sequencing,MPS)或深度测序,基于具有广泛功能的多步核酸的技术对已知和未知外源因子进行不可知检测。在某些情况下,NGS 可用于靶向检测已知病毒。
平台制造(根据 ICH Q11)
制定新药的制造策略方式,所用的生产工艺与同一申办人在生产相同类型的其他药物相似(例如:在单克隆抗体的生产中,使用预先确定的宿主细胞、细胞培养和已有丰富经验的纯化工艺)。
平台验证
在本指导原则中,该术语仅指病毒清除的平台验证。
由此而论,平台验证定义为使用其他产品的先验知识(包括病毒降低水平数据的内部经验),以便根据目前的理解说明新的类似产品的下降因子情况。
先验知识
先验知识是指现有知识,包括内部知识(如开发和生产经验)、外部知识(如科学和技术出版物,包括供应商数据、文献和同行评审的出版物)或既定科学原理(如化学、物理和工程原理)的应用。
病毒清除工艺鉴定
病毒清除研究中,用非特异性“模型”病毒评价生产工艺去除和/或灭活病毒稳健性。
病毒清除工艺验证研究
病毒清除研究中,用“相关”和/或特异性“模型”病毒确定生产工艺去除和/或灭活这些病毒的能力。
病毒清除工艺的稳健性
术语稳健性用于描述两种不同特征之一。其一是工艺或工艺步骤耐受材料变化和工艺变更而不对病毒清除产生负面影响的能力。另一个特征是清除各种特异和非特异性模型病毒的能力。
生产用细胞
用于生产产品的细胞基质。
补充检测方法
用于提供数据以完善常规检测的试验方法。是用于克服现有检测方法局限性(例如供试品干扰或毒性)的试验方法。
未加工收获液
一次或多次收获后汇集起来的细胞和培养基。当细胞不能直接获得时,未加工收获液是从培养罐中收获的液体。
病毒
在细胞内复制并有潜在致病性的感染性因子,只有一种核酸(RNA 或 DNA),不能以其遗传物质形式生长、二分裂和繁殖。
- 外源性病毒
- 非故意引入的污染病毒。
- 内源性病毒
- 其基因组是细胞系来源物种种系的一部分,并共价整合到衍生亲本细胞系的动物基因组中的一类病毒体。在本指导原则中,故意引入的非整合病毒,如用于永生化细胞基质的 EB 病毒或牛乳头瘤病毒。
- 辅助病毒
- 在本指导原则中,辅助病毒是指能够促进产品表达或复制的病毒或病毒载体。
- 非特异性模型病毒
- 用于鉴定生产工艺清除病毒能力的病毒,其目的是对生产过程去除和/或灭活病毒的总体能力进行鉴定(即鉴定纯化工艺的稳健性)。
- 相关病毒
- 工艺评价研究用病毒,可以是己鉴定的病毒或是己知病毒的同一种类,或是生产过程中使用的任何易污染细胞培养基质或其他生产用试剂或材料的病毒。
- 特异性模型病毒
- 与己知或可疑病毒密切相关(同种或同属)的病毒,与所观察到的或可疑的病毒具有类似的理化特性。
病毒清除
通过去除病毒颗粒或灭活病毒感染性的方法消除目标病毒。
病毒样颗粒
在电镜下可见结构,形态与己知病毒相似。
病毒去除
用物理方法将病毒颗粒从预期产品中分离。
病毒载体
一种重组病毒,可在体内作为药品应用或在体外应用于其他先进治疗。基因工程病毒载体的生产可能需要辅助病毒。
病毒载体衍生产品
由重组病毒编码和表达的产品。基因工程病毒载体的生产可能需要辅助病毒。
工作细胞库(WCB)
在规定培养条件下扩增 MCB 获得匀质的细胞悬液,并经分装成多个等份制备而成。
工作病毒种子(WVS)
工作病毒种子(批次或库)产生于 MVS。
| MCB | WCBa | 达到 LIVCA 的细胞 b | |
| 逆转录病毒和其他内源性病毒检测 | |||
| 感染性 | + | – | + |
| 电镜c | +c | – | +c |
| 逆转录酶d | +d | – | +d |
| 其他特异性病毒检测e | 如适用e | – | 如适用e |
| 非内源或外源性病毒检测 | |||
| 体外试验或NGSj | +f | +f | +f |
| 体内试验或NGSj | +g | –g | +g |
| 抗体产生试验或特异性分子试验 h,j | +h | – | – |
| 其他特异性病毒检测i | +i | – | – |
a. 第 3.1.2 节。
b. 代次限制细胞:在体外达到传代限次的生产用细胞(见第 3.1.3节)。
c. 也可测定其他因子。
d. 如果已知细胞系能结构性地产生逆转录病毒颗粒,则无需进行检测。
e. 适用于己知受此类因子感染的细胞系。
f. 可直接对 WCB 或直接来源于该 WCB 的 LIVCA 细胞进行体外病毒检测。根据风险评估,使用广义分子生物学方法(NGS)进行的病毒检测可作为体外检测(细胞培养和 PCR)的补充或替换试验。
g. 可根据风险评估进行体内试验。然而,基于细胞系历史、先验知识和其他基于风险的考虑,无需对充分表征的细胞系(如CHO、NS0 和 SP2/0)进行体内试验。包括事先对亲本未转染细胞系进行体内病毒检测或 NGS 检测,以及对来源于亲本细胞库的 MCB 进行的控制。还应考虑对来自同一亲本细胞库的其他 MCB 进行病毒安全性检测的先验知识,包括用于建立 MCB 的方法。如果第一个 WCB 或后续 WCB 在批准的受控条件下制备,则通常不需要进行检测。对于处于 LIVCA 中的细胞,根据先验知识和其他基于风险的考虑,可能不需要进行检测。如果仍有风险,可考虑保留检测或用分子生物学方法(如 NGS或 PCR)替代进行广泛的病毒检测,以检测 MCB 构建期间或 LIVCA 阶段细胞培养期间可能带入的病毒。
h. 例如 MAP、RAP、HAP——通常适用于啮齿细胞系。病毒特异性 PCR 或靶向分子生物学方法可用作替代检测法进行动物试验。例如,基于细胞系的来源和历史,包括相关原材料和试剂。
i. 例如,基于细胞系的来源和历史,包括相关原材料和试剂。
j. 根据试验适用性和风险评估,当适用时,应考虑用 NGS 法替代体内检测法,并可用其补充或替代体外和其他病毒特异性试验。
| 检测项目 | 供试品 | 检测能力 | 检测局限性 |
| 抗体产生试验 | 细胞裂解物及其培养基 | 特异病毒抗原 | 对动物试验体系抗原无感染性 |
| 体内病毒筛查 | 细胞裂解物及其培养基 | 各种病毒 | 在试验体系中病毒无法复制或致病 |
| 体外病毒筛查:1. 细胞库检定 2. 生产筛查 | 1. 细胞裂解物及其培养基(共培养时,供试品中的细胞应是完整的细胞) 2. 从生产反应器中收获的未加工收获液或细胞裂解物及其培养基 | 各种病毒 | 在试验体系中病毒无法复制或致病 |
| TEM 检测:1. 细胞基质 2. 细胞培养上清液 | 1. 活细胞2. 无细胞培养上清液 | 病毒或病毒样颗粒 | 在鉴别的同时进行定性分析 |
| 逆转录酶(RT) | 无细胞培养上清液 | 逆转录病毒和己表达的逆转录病毒RT | 只能测定在理想条件下具有最佳活性的酶。很难解释是由于细胞中酶的存在还是浓缩样本的背景干扰 |
| 逆转录病毒(RV)感染性 | 无细胞培养上清液 | 感染性逆转录病毒 | RV 在所选试验体系中不能复制或形成离散点或菌斑 |
| 共培养1. 感染终点 2. TEM 终点 3. RT 终点 | 活细胞 | 感染性逆转录病毒 | RV 不能复制1. 见上文 RV 感染性栏 2. 见上文 TEMa 栏 3. 见上文RT 栏 |
| PCR(聚合酶链反应) | 细胞、培养液和其他材料 | 特异性病毒序列 | 必须有引物序列。不能说明病毒是否有感染性。 |
| NGS | 细胞、培养液和其他材料 | 各种病毒 | 阳性结果并不能说明病毒是否有感染性,可能需要进一步研究 |
a.此外,可能很难将供试品与指示细胞区分
| MAP4 | HAP4 | RAP4 |
| 鼠痘病毒2,3 | 淋巴细胞性脉络丛脑膜炎病毒(LCM)1,3 | 汉坦病毒1,3 |
| 汉坦病毒1,3 | 小鼠肺炎病毒 (PVM)2,3 | 基尔曼氏大鼠病毒 (KRV)2,3 |
| K 病毒 2 | 呼肠孤病毒 3 型 (Reo3)1,3 | 小鼠脑脊髓炎病毒 (Theilers, GDVII)2 |
| 乳酸脱氢酶病毒 (LDM)1,3淋巴细胞性脉络丛脑膜炎病毒 (LCM)1,3, 小鼠细小病毒 2,3 小鼠腺病毒 (MAV)2,3 小鼠巨细胞病毒 (MCMV)2,3 | 仙台病毒 (SV)1,3SV5 | 小鼠肺炎病毒 (PVM)2,3大鼠冠状病毒 (RCV)2 呼肠孤病毒 3 型 (Reo3)1,3 仙台病毒1,3 涎泪腺炎病毒 (SDAV)2 |
| 小鼠脑脊髓炎病毒 (Theilers, GDVII)2 | 杜兰氏 H-1 病毒 2,3 | |
| 小鼠肝炎病毒 (MHV)2小鼠轮状病毒 (EDIM)2,3 | ||
| 小鼠肺炎病毒 (PVM)2,3 | ||
| 多瘤病毒 2 | ||
| 呼肠孤病毒 3 型 (Reo3)1,3仙台病毒1,3 | ||
| 胸腺病毒2 |
1. 有证据表明能感染人或灵长类的病毒。
2. 无证据表明能感染人的病毒。
3. 能在人或灵长类来源的细胞中体外进行复制的病毒。
4. NAT(如 PCR 检测法或其他靶向分子生物学方法)可用于替代特定啮齿类动物病毒检测。
| 情况A | 情况B | 情况C2 | 情况D2 | 情况E2 | 情况F | |
| 状态 | ||||||
| 存在病毒 1 | – | – | + | + | (+)3 | – |
| 病毒样颗粒 1 | – | – | – | – | (+)3 | – |
| 逆转录病毒样颗粒 1 | – | + | – | – | (+)3 | – |
| 己鉴定病毒 | 不适用 | + | + | + | – | + |
| 对人有感染性的病毒 | 不适用 | –4 | –4 | + | 未知 | (+)9 |
| 存在辅助病毒 | – | – | – | – | – | + |
| 行动 | ||||||
| 用非特异性“模型” 病毒对病毒清除进行工艺表征 | 是 5 | 是 5 | 是 5 | 是 5 | 是 7 | 是 5 |
| 用“相关”或特异性“模型”病毒对清除病毒工艺进行评价 | 否 | 是 6 | 是 6 | 是 6 | 是 7 | 是 9 |
| 检测纯化后产品中的病毒 | 不适用 | 否 | 是 8 | 是 8 | 是 8 | 是 9 |
1. 细胞基质和/或在未加工收获液阶段病毒检测结果。除非经过特定的病毒清除和风险评估,否则一般不应使用被病毒污染的细胞培养物进行生产。内源性病毒(如逆转录病毒)或是 MCB 组成部分的病毒,在经过适当的病毒清除评价后可接受。
2. 当源材料受到病毒污染时,无论其是否已知对人类具有传染性和/或致病性,将只能在极个别情况下经过特定的病毒清除证明和风险评估后才被允许使用。
3. 用直接或间接的方法观察过的病毒。
4. 被认为对人无致病作用。
5. 应使用非特异性“模型”病毒对病毒清除进行表征。
6. 应使用“相关”或特异性“模型”病毒进行工艺评价。
7. 见正文中情况 E。
8. 应对可疑病毒使用适当的具有高特异性和高灵敏度的方法进行检测,确认纯化后产品中无可检测到的病毒。上市审批时,应至少提供三批试生产规模或生产规模的纯化后产品的检定数据。
9. 病毒可能对人类致病,也可能不致病。应对辅助病毒(重组或野生型)进行工艺评价。如果不可行,则应使用特异性模型病毒。当用于生产时,在未加工收获液阶段,针对至少三个细胞培养活动的辅助病毒进行定量检定,以确定病毒清除的目标。纯化后,针对相关允许细胞系使用感染性试验进行病毒敏感性检测,确认无可检测到的辅助病毒。此外,也可使用分子生物学方法进行检测。应确认每个纯化后产品无残留辅助病毒。
附件 1:已鉴定细胞库经体内培养表达的产品
对于从接种已鉴定细胞库细胞的动物中收获的液体提取的产品,应另外提供有关该动物的资料。
用于制造生物技术产品/生物制品的动物应尽可能地从种类明确而又无特异病原体的动物群中选择。应对表 3 所列的有关病毒进行充分检测。应对新进及已患病动物的检疫程序说明,并确保设施内采用的所有容器、清洁和去污方法足以控制外源因子的传播。这可通过一项警戒程序加以实现。还应列出一份待检测外源因子清单。兽医服务应现场或就近提供。应说明动物饲养场与其他生产设施隔离的情况。人员操作应确保安全。
应充分说明动物饲养的程序。其中包括:饮食、清洁以及喂养计划、定期的兽医检查规定(若适用),以及动物接种后可能需要的特殊处理的细节。同时要对动物的接种方案、接种物的制备、接种部位及接种途径加以说明。
来源于动物的原始收获物可看做是从生物反应器中收获的未加工收获液。因此,本文件上文第 4 节所列的所有检测项目都应适用。此外,制造商应对未加工收获液的生物负荷进行评估,确定该材料是否无支原体,同时对成年和哺乳期小鼠进行种属特异性检测及体内测试。
附件 2:病毒清除研究用病毒的选择
A. 有效的“模型”病毒举例
a. 代表一定理化结构的非特异性“模型”病毒:
- – SV40(多瘤病毒)、动物细小病毒或其他一些小的非脂包膜病毒;
- – 副流感病毒或流感病毒、辛德比斯(Sindbis)病毒或其他一些中到大型脂包膜 RNA 病毒;
- – 疱疹病毒(如单纯疱疹病毒( HSV-1 )或伪狂犬病毒(PRV))或其他一些中到大型 DNA 病毒。以上所列仅为一些病毒的示例,不强制使用。
b. 对于产生逆转录病毒样颗粒的细胞基质,鼠逆转录病毒常用作特异性“模型”病毒。也可以使用内源性鼠或其他啮齿动物逆转录病毒颗粒。
B. 已用于病毒清除研究的病毒举例
表 A-1 列举了己用于病毒清除研究的一些病毒。但是,由于其仅仅是示例,对表中任何病毒的使用均为非强制性规定。制造商也可考虑使用其他病毒,特别是对其自身生产工艺更适合的病毒。一般说来,至少要用三种不同特性的病毒对工艺的清除病毒能力进行评估。
| 病毒 | 科 | 属 | 天然宿主 | 基因组 | 包膜 | 大小(nm) | 形状 | 耐受性 a |
| 水疱性口炎病毒b | 弹状病毒 | 水泡性病毒 | 马牛 | RNA | 有 | 70×150 | 子弹状 | 低 |
| 副流感病毒 | 副粘病毒 | 副粘病毒 | 多种 | RNA | 有 | 100-200+ | 多形/球形 | 低 |
| MuLV | 逆转录 | γ 逆转录病毒 | 小鼠 | RNA | 有 | 80-110 | 球形 | 低 |
| Sindbis 病毒 | 披膜病毒 | 甲病毒 | 人 | RNA | 有 | 60-70 | 球形 | 低 |
| BVDV | 黄病毒 | 瘟病毒 | 牛 | RNA | 有 | 50-70 | 多形-圆形 | 低 |
| 伪狂犬病毒b,c | 疱疹病毒 | 水痘病毒 | 猪 | DNA | 有 | 120-200 | 球形 | 中 |
| 苜蓿银纹夜蛾核型多角体病毒c | 杆状病毒 | α 杆状病毒 | 昆虫 | DNA | 有 | 250-300 | 多面体 | 中 |
| 腺病毒 2 型或 5 型c | 腺病毒 | 腺病毒 | 人 | DNA | 无 | 70-90 | 二十面体 | 中 |
| 水疮性病毒 2711 | 杯状病毒 | 水疮性病毒 | RNA | 无 | 27-40 | 二十面体 | 中 | |
| 脑心肌炎病毒(EMCV) | 小 RNA 病毒 | 心病毒 | 小鼠 | RNA | 无 | 25-30 | 二十面体 | 中 |
| 呼肠孤病毒 3 型 | 呼肠孤病毒 | 正呼肠孤病毒 | 多种 | RNA | 无 | 60-80 | 球形 | 中 |
| SV40 | 多瘤病毒 | 多瘤病毒 | 猴 | DNA | 无 | 40-50 | 二十面体 | 很高 |
| 细小病毒(犬/鼠/猪)d | 细小病毒 | 细小病毒 | 犬、鼠、猪 | DNA | 无 | 18-24 | 二十面体 | 很高 |
a:指生产工艺研究中,对理化处理具有的耐受性。耐受性与特定的处理有关,只有在了解病毒生物学特性和生产工艺性质的情况下才能使用。实际结果会随着处理情况而变化。
b:在昆虫细胞中发现杆状病毒的相关模型
c:用于病毒载体生产的辅助病毒的特异性模型或相关病毒
d:在验证病毒过滤器时,可用作较大球形/二十面体病毒和脂包膜病毒的单个最坏情况模型病毒。
以上所列病毒仅为示例,不强制使用。
附件 3:评估病毒和病毒下降因子的统计学考虑
所有生物学检测方法在测定病毒滴度时都会碰到变异的问题。应对病毒滴定和从中获得的下降因子的准确性以及检测方法的有效性进行评估,从而确定研究的可靠性。统计学评价的目的是确认病毒清除研究工作是在病毒学可接受水平上进行的。
1. 检测方法可以是定性的或是定量的。定性法包括动物感染性检测或组织培养感染剂量(TCID)测定,该法是按动物或细胞培养感染与否来记分。然后根据动物或培养物感染的比例测定感染滴度。在定量分析方法中,所测定的感染性随病毒的不断加入而变化。定量法包括基于分子的方法或空斑检测,即计算每一感染单位的相应空斑。定性和定量分析都可进行统计学评估。
2. 变异可来自稀释误差、统计效应和检测体系中未知的或难以控制的差异。不同检测运行间的变异(试验间变异)将大于同一检测运行间的变异(试验内变异)。
3. 试验内变异的 95%置信限一般应在均值±0.5 log10 范围内。试验内变异可用教科书上的标准方法进行评估。试验间变异可引入参考品进行监测,其效价的估计值应在实验室平均估计值的 0.5 log10 左右(试验可接受)。如有正当理由,较低精密度的分析方法也是可以接受的。
4. 只要可能,在进行“相关”和特异性“模型”病毒清除研究时,应计算所观察到的下降因子的 95%可信限。如果起始材料病毒测定的 95%可信限是+s,此后的材料病毒测定 95%可信限是+a,下降因子的 95%可信限为:
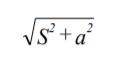
低浓度病毒检测的概率
显然,在低病毒浓度时(如每升含 10-1000 个感染性颗粒),几毫升样本中可能含有也可能不含感染性颗粒。该样本不含感染颗粒的概率 p 为:
p=((V-v)/V)n
其中 V(升)是待检测材料的总体积,v(升)是样本体积,n 是在 V 中统计学分布的感染颗粒的绝对数目。
如果V>>v,这一公式可用泊松分布近似计算:
p=e-cv
其中c 为每升感染性颗粒浓度。
或:c=ln p/-v
举例: 如果测试样本体积为 1 毫升, 在病毒浓度为每升 10~1000 个感染性颗粒,则概率 p 为:
| c | 10 | 100 | 1000 |
| p | 0.99 | 0.90 | 0.37 |
这说明,对于每升含 1,000 个病毒颗粒浓度的样本,抽样的37%中,其 1 ml 样本中不含一个病毒颗粒。
如果仅对样本的一部分进行病毒检测,并且检测结果为阴性,则应对总样本中存在的病毒数量进行计算,以获取阳性结果,并在计算下降因子时将此值考虑在内。应达到 95%可信限。但是,在某些情况下,由于材料的局限性,这一操作可能不实际。
附件 4:确定病毒清除率研究中下降因子的计算
单个纯化或灭活步骤的病毒下降因子是指纯化前样本中病毒载量与用以下一步工艺的纯化后样本中病毒载量之比的 log10。如使用以下缩写:
起始物料:
体积v’;滴度 10a’;
病毒载量:(v’)(10a’),
最终材料:
体积v;滴度 10a“;
病毒载量:(v”)(10a</sup”),
单个下降因子 Ri 按如下公式计算:
10Ri=(v’)(10a’)/(v”)(10a”)
这一公式同时考虑了纯化前后样本的滴度和体积。
由于有些病毒滴定本身不精确,用于计算总下降因子的单个下降因子应大于 1。
整个生产过程的总下降因子是各单个阶段下降因子对数之和。它代表了第一个清除工艺开始时的病毒载量与最后一步清除工艺结束时的病毒载量之比的对数。下降因子一般用 log 表示,说明尽管残留的病毒感染性可能己大大降低,但是决不会降至 0。
附件 5:每剂量病毒颗粒的估算
本附件适用于可估计起始数量的病毒,如内源性逆转录病毒。举例:
I. 假设
细胞培养收获液中,测得或估计的病毒浓度=106/ml
计算得病毒清除因子=>1015
一个剂量的产品所需的培养收获液体积=1 升(103 ml)
II. 每剂量病毒颗粒的估算
[(106 病毒颗粒/ml)×(103 ml/剂量)]/ (清除因子>1015)
=(109 病毒颗粒/剂量) / (清除因子>1015)
=<10-6 颗粒/剂量 1
因此,可以预计每一百万剂量中的病毒颗粒少于一个。
上述情况是采用啮齿类动物细胞生产单克隆抗体期间内源性逆转录病毒减少的典型情况(情况 B)。在针对特异性病毒的综合风险评估中,应考虑其他因素,如病毒的宿主范围、病毒的致病性、避免污染的措施、检测措施、给药途径和人类感染剂量。
在中国仓鼠卵巢(CHO)细胞的情况 B 中,如果体外检测未能鉴别出感染性逆转录病毒的存在,则认为重组蛋白的逆转录病毒样颗粒(RVLP)的安全限度(<10-4 个颗粒/剂量)可接受。
附件 6:应用先验知识(包括内部经验)以减少特定产品验证工作的举例
根据平台验证方法的一般原则,需要证明来自同一平台的产品具有稳健的病毒清除能力,而且病毒清除的程序需遵循既定且经充分鉴定的条件。此外,应证明产品中间体成分与病毒清除研究中使用的中间体类似,除非先验知识表明病毒清除工艺对产品中间体成分具有稳健性。
由此而论,平台验证定义为使用其他产品的先验知识,包括病毒降低数据的内部经验(申请人拥有的数据),说明新的类似产品的下降因子情况。一般而言,基于先验知识(包括内部经验)的新产品病毒清除声明应包括对所有可用数据的讨论以及支持平台验证方法的基本原理(参见第 6.6 节)。用于减少产品特定验证的部分先验知识和内部数据可以作为新产品及其生产工艺与其他内部产品、相关工艺条件和产品中间体的比较予以提供。
专门用于清除病毒的工艺步骤,如通过洗涤剂、低 pH 值 灭活以及通过病毒过滤去除,适用于平台验证方法。
因此,以下内容给出了通过洗涤剂、低 pH 值孵育以及病毒过滤对XMuLV 进行灭活/去除的先验知识应用示例。
所示模拟示例仅用于说明目的,表明如何应用平台验证方法,而不用作监管提交的模板或唯一依据。
基于当前对整个行业应用的各种工艺条件的理解,工艺参数及其对各个工艺步骤的潜在重要性汇总见表 A-2 至 A-4。应根据先验知识和内部经验评估工艺参数和中间体对 X-MuLV 清除的实际影响。
基于工艺理解不断发展,未来可能会推荐更多的工艺步骤用于平台验证。
使用溶剂/洗涤剂(SD)或洗涤剂单独灭活
基于作用机制,SD 试剂的洗涤剂浓度或单独的洗涤剂浓度是重要的工艺参数。
此外,疏水性杂质(如脂质、细胞碎片或细胞培养基的成分(如消泡剂))可通过激发洗涤剂或 SD 混合物溶解病毒脂质包膜而影响病毒灭活,因此应进行评估。
迄今为止,尚未有迹象表明病毒与特定治疗性蛋白之间的相互作用会影响洗涤剂的灭活作用。聚集体(例如,细胞碎片或聚集的病毒颗粒)可能会包埋病毒颗粒并防止其与洗涤剂接触。因此在生产时,产品中间体,例如收获的细胞培养液(HCCF),应从细胞/细胞碎片中澄清,包括在洗涤剂灭活之前进行一步≤0.2 µm 标称孔径的过滤。
以下段落以 SD 或 Triton X-100 为例,描述了如何将平台验证方法应用于 XMuLV 灭活。该方法也可能适用于已证明可强而有效进行XMuLV 灭活的替代洗涤剂。
Triton X-100 是一种非离子洗涤剂,常用于溶解脂质双层的膜研究。其通过溶解病毒脂质包膜来灭活脂包膜病毒,从而使病毒失去感染性。多年来,Triton X-100 已广泛用于血浆衍生产品生产过程中的病毒灭活,以及添加到 HCCF 中用于单克隆抗体(MAb)平台纯化过程中的病毒灭活。
由于环境中降解化合物具有类激素活性,欧洲化学品管理署将Triton X-100 列入授权清单(附件 XIV)。因此,尽管其被广泛使用,制药行业正在寻找可替代洗涤剂。其他具有类似理化特性的洗涤剂可于市场上购买,其也可实现有效的 XMuLV 灭活。
由于 Triton X-100 的非离子性质,其有效性不应受 pH 值、离子强度或 HCCF 中反离子性质的影响。既往经验表明,在 0.2% Triton X-100 浓度、15℃ 和 60 分钟的孵育条件下,来自平台工艺的多种产品均可有效灭活 HCFF 中的 XMuLV, HCCF 涵盖一系列典型脂质和总蛋白含量。然而,如下所述,在不进行特定产品实验时,建议应用 0.5%的Triton 浓度,以确保有效和可靠的灭活。
基于洗涤剂的脂质包膜病毒灭活的工艺参数及其潜在重要性汇总如表 A-2 所示。
| 工艺参数 | 潜在影响 | 依据 |
| SD 或 Triton X-100 浓度 | 高 | 灭活剂 |
| 孵育时间 | 高 | 灭活机制具有时间依赖性 |
| 温度 | 高 | 对灭活动力学的影响 |
| 经 0.2 µm 过滤进行预处理 | 高 | 从起始中间体中去除可能会包埋病毒颗粒并防止其与洗涤剂接触的聚集体 |
| HCCF 中的总脂质含量或替代参数 | 低 | 在最坏情况HCCF 下观察到低影响 |
| 产品类型 | 低 | 未观察到对 MAb、半抗体、融合蛋白或重组蛋白的灭活影响 |
| HCCF 中的总蛋白含量 | 低 | 在最坏情况HCCF 下观察到低影响 |
| pH | 低 | Triton X-100 是一种非离子洗涤剂 |
| 离子强度 | 低 | 见上文 |
| HCCF 中的缓冲盐 | 低 | 见上文 |
| 病毒颗粒与产品之间的潜在相互作用 | 低 | 未观察到对灭活的影响,破坏脂质包膜可降低与产品相互作用的概率 |
因此,基于目前的工艺理解,在≥15°C 条件下使用≥0.5% Triton X-100 对澄清的 HCCF 进行≥60 分钟的处理,可有效灭活用于多种细胞培养衍生产品的 XMuLV。使用 1%的 Triton X-100 和 0.3%的磷酸三丁酯(TNPB)处理≥30 分钟,或在≥23℃条件下使用 1%的聚山梨酯 80 和 0.3%的 TNBP 处理≥6 小时,可有效灭活逆转录病毒。根据目前的工艺理解,平台验证方法可应用于通过 SD 处理或单独使用Triton X-100 处理的XMuLV 灭活。
低 pH 孵育
低 pH 处理是通过使病毒包膜中的蛋白质变性,从而破坏脂质包膜来灭活脂包膜病毒。捕获色谱后工艺中间品的低 pH 处理已广泛用于细胞培养衍生产品(如单克隆抗体(MAb))生产过程中的逆转录病毒灭活。
灭活效率取决于作为灭活剂的氢离子浓度,以 pH 值、孵育时间和温度以及缓冲液基质来衡量。极高的离子强度也可能影响灭活效率。
工艺参数及其对低 pH 灭活 XMuLV 的潜在影响汇总如表 A-3所示。
| 工艺参数 | 潜在影响 | 依据 |
| pH | 高 | 灭活剂 |
| 孵育时间 | 高 | 灭活机制具有时间依赖性 |
| 温度 | 高 | 对灭活动力学的影响 |
| 缓冲液基质 | 高 | 现有数据显示,灭活稳健性取决于缓冲液基质 |
| 产品浓度 | 低 | 未观察到对灭活的影响 |
| 产品类型 | 低 | 未观察到对 MAb、半抗体、双特异性抗体、融合蛋白或重组蛋白的灭活影响 |
| NaCl 浓度(a) | 低 | 如果氯化钠≤500 mmol/L,则无影响 |
| 病毒颗粒与产品之间的潜在相互作用 | 低 | 未观察到对灭活的影响。 |
(a):迄今为止,关于其他缓冲液离子强度影响的数据有限。
基于目前的工艺理解,在≤pH 3.6、≥15°C、≤500 mmol/L 氯化钠浓度下,≥30 分钟的低pH 处理可有效灭活 XMuLV。乙酸盐和枸橼酸盐缓冲液是最常用的缓冲液,可有效灭活 XMuLV。
根据目前的工艺理解,平台验证方法可应用于低 pH 处理的 XMuLV 灭活。
病毒过滤
病毒过滤的作用机理是基于粒径的病毒颗粒去除。一般而言,产品中间体的体积通量以及冲洗过滤器的缓冲液的体积通量和压力(包括压力中断)是病毒过滤的潜在关键参数。
在病毒粒径远远大于过滤器孔径分布的情况下,病毒颗粒与产品的潜在相互作用并不重要。然而,在病毒粒径和孔径相似的情况下,尚未完全了解潜在相互作用对流体动力学和病毒截留的影响。本节重点介绍利用其他产品在病毒过滤方面的先验知识和内部经验,说明通过小型和大型病毒截留过滤器去除逆转录病毒的相关信息。
影响小型病毒过滤器有效去除逆转录病毒的因素已耳熟能详,如膜类型、流速或压力受控的过滤模式和压力中断等工艺参数的变化。基于病毒去除的可预测性和稳健性,该工艺步骤适用于平台验证方法。
针对使用小型病毒过滤器进行的病毒去除,一种选择是对较大的球形/二十面体病毒和脂包膜病毒应用细小病毒的对数下降值。 然而,这可能导致因细小病毒通过而低估病毒清除能力(例如逆转录病毒清除能力)。鉴于基于粒径的作用机理,以及使用小型病毒过滤器彻底去除逆转录病毒的行业经验,公司可以使用其从细小病毒和逆转录病毒去除中获得的内部数据,为常用小型病毒过滤器建立平台逆转录病毒清除声明。
根据基于粒径的去除机理,理论上对小型病毒通过小型病毒截留过滤器的病毒传代风险比逆转录病毒高。
需要透彻了理解压力中断的影响,以及保留反映 GMP 条件下的体积通量和过滤器冲洗量。
如果利用其他产品的先验知识和内部经验来声明细小病毒的去除情况,则至少需要使用细小病毒进行一次特定产品的确认验证。
病毒过滤器的类型对于减少病毒及其在工艺参数影响方面的稳健性非常重要,应在设计平台数据时纳入考量。
| 工艺参数 | 潜在影响 | 依据 |
| 病毒过滤器加载产品中间体的体积通量 | 高 | 已观察到低水平细小病毒通过(取决于具体的过滤器类型) |
| 冲洗过滤器用缓冲液的体积通量 | 高 | 已观察到低水平细小病毒通过 |
| 压力 | 高 | 压力不应超过过滤器运行的上限。对于特定的膜类型,低压可能会出现更坏的情况。应考虑到压力中断(如果在过滤期间或从产品中间体过滤切换到过滤器冲洗时发生)情况。 |
| 产品类型 | 低 | 未观察到对 MAb、半抗体、双特异性抗体、融合蛋白或重组蛋白的病毒清除影响 |
| 产品浓度 | 低 | 未观察到对病毒清除的负面影响 |
| pH | 低 | 未观察到基于粒径的去除对病毒清除的负面影响 |
| 离子强度 | 低 | 观察到对病毒清除的影响有限 |
| 缓冲液基质 | 低 | 观察到对病毒清除的影响有限 |
| 病毒颗粒与产品之间的潜在相互作用 | 低 | 病毒与抗体之间的特异性相互作用可增强病毒截留 |
附件 7:基因工程病毒载体和病毒载体衍生产品
7.1 介绍
生物技术的进步导致新的和先进的生产平台的出现,用以呈现使用人或动物来源的已表征细胞库(即鸟类、哺乳动物或昆虫)生产的新产品类型。附件 7 的范围包括辅助病毒依赖性和辅助病毒非依赖性的基因工程病毒载体以及病毒载体衍生产品,根据对产品理化特性的考虑,这些产品适合进行病毒清除。这些产品包括病毒样颗粒(VLP)和蛋白质亚基,例如使用杆状病毒/昆虫细胞生产的产品、基于纳米颗粒的疫苗以及病毒载体产品(如 AAV)。这些药品可应用于体内或体外。
辅助病毒非依赖性产品是使用稳定转化或瞬时转染的细胞系,或通过感染蛋白表达病毒载体(例如重组杆状病毒)生产。辅助病毒依赖性产品需要辅助病毒来使产品表达或病毒载体复制,例如腺相关病毒或使用辅助病毒如单纯疱疹病毒或腺病毒表达的重组蛋白。
生物制药产品病毒污染的潜在来源如主要指导原则第 2 节中所述。应考虑额外的污染风险,例如由表达系统引入的风险,以及复制型病毒污染的可能性。在评估产品生产过程中外来污染的可能性时,应仔细考虑细胞基质对外源病毒的易感性。使用充分表征的细胞库和病毒种子可降低病毒污染的风险。此外,用于生产的辅助病毒被视为工艺相关病毒污染物。
新产品类型的病毒安全性和污染控制可通过材料采购的综合方案的应用、在适当的生产步骤进行病毒检测以及通过生产工艺去除和/或灭活外源病毒和辅助病毒得到保证。在病毒清除受限的情况下,病毒安全性应集中在原材料和试剂以及生产工艺的检测和控制方面。
因此,应采用基于风险的方法来证明产品的病毒安全性。
7.2 病毒检测
应在适用的生产阶段对内源和外源性病毒污染进行大量检测和特性鉴定,以支持产品的整体安全性。基于产品类型及其相关风险因素,检测方案应适用于整个产品生命周期。在生产过程中各个工艺步骤所要进行的检测概述如下表 A-5 所示。对适用于病毒种子、载体收获物和原液的检测进行了描述。虽然用于病毒载体生产的细胞基质所需的检测和特性鉴定方案与主要指南文件中的表 1 大体一致,但针对这些产品类型可能还需要考虑其他方面信息,因此为了完整起见,在下文表 A-5 中具体说明。
检测类型和程度取决于风险评估,且应考虑与细胞基质和生产工艺相关的特定风险因素。应考虑的因素包括细胞基质和病毒载体的来源、传代历史和特性鉴定、所用的原材料、试剂和培养方法、对辅助病毒的依赖性以及生产工艺灭活和/或去除病毒的能力。
| 检测 | MCB、WCB、 达到 LIVCA 的细胞 | 病毒种子k | 未加工收获液 | 原液 |
| 检测外源或内源病毒 | ||||
| a, b, 体外检测或 NGS | i, 见主要指导原则表 1 | +h | +h | – |
| b, 体内检测或NGS | + h | – h,l | – | |
| c 其他特异病毒检测 | l | l | – | |
| d 抗体产生试验或特异性分子试验 | +j,l | – | – | |
| 内源性、辅助和复制型病毒的检测(如适用) | ||||
| e 逆转录病毒 | i, 见主要指导原则表 1 | + | +l | – |
| f 残留辅助病毒 | NA | – | + | + l |
| g 复制型病毒 | + | + | (+) | (+) |
a 应根据风险评估对允许细胞系进行检测。应观察指示细胞培养物至少两周,并对二次传代进行两周的进一步观察。包括红细胞吸附和红细胞凝集病毒检测。对于在昆虫细胞系中生产的产品,应包括用于检测虫媒病毒的允许细胞系(例如 BHK 细胞)。如果病毒载体和病毒载体衍生产品无法被中和,则可以使用经验证的替代试验。在进行下游加工之前,应对病毒种子和未加工收获液进行检测。在某些情况下,未加工收获液可能与原液相同。
b 如适用,应考虑使用广义 NGS 替代体内外源性病毒检测,并可根据试验适用性和风险评估用于补充或替代体外检测。
c 基于细胞基质、原材料或试剂以及生产工艺的风险评估,确定种属特异性病毒检测法(例如,NAT 和细胞培养或靶向 NGS)。如果使用血清成分或胰蛋白酶,则可能包括人类或啮齿类动物种属特异性病毒、昆虫细胞中的虫媒病毒以及牛或猪的病毒。
d 基于细胞基质、原材料或试剂以及生产工艺的风险评估,进行抗体产生试验( MAP、HAP 、RAP)或特异性病毒 NAT 或靶向 NGS。
e 应考虑在 MCB 和病毒种子阶段使用逆转录酶活性测定检查是否存在逆转录病毒。如果 MCB 或病毒种子的逆转录酶活性为阳性,则接下来应对来自至少 3 个细胞培养活动的未加工收获液进行包括定量的潜在逆转录病毒颗粒检定,以确定病毒清除的目标水平。此外,应根据风险评估,采用基于 PCR 的 RT 测定法(PBRT)对未加工收获液进行检测,例如,产物增强型逆转录酶活性测定法(PERT)。
f 当用于生产时,应在未加工收获液阶段使用至少 3 个细胞培养活动对辅助病毒进行量化,以确定病毒清除的目标。纯化后,应针对相关允许细胞系使用感染性试验进行病毒敏感性检测,确定无可检测到的辅助病毒。此外,也可使用分子生物学方法进行检测。应针对每个纯化后产品确认其无残留辅助病毒(情况 F,表 4)。
g 复制型病毒(RCV)可在生产过程中的任何步骤(例如,在初始转染或转导步骤以及整个生产过程中)产生。目前的建议包括在生产的多个阶段检测 RCV,以检测重组或载体病毒是否回复至亲本或野生型表型。检测所适用的检定阶段和方法应根据产品特性而定。例如,在病毒种子或细胞库鉴定期间,对来自稳定转染载体产品或 MCB 和 LIVCA 的细胞和上清液进行 RCV 检测。RCV 检测适用于生产期间,适用时对各未加工收获液或各原液/最终批次的载体生产细胞和上清液进行检测。例如,通常在未加工收获液中进行复制型病毒检测,以确保基于产品的腺相关病毒(AAV)的可检测性或原液步骤的可行性,在表中以+/-表示。
h 在病毒种子和未加工收获液阶段检测平行培养的对照细胞,可能发生试验干扰。
i 对于昆虫来源的细胞系,应进行种属特异性病毒和虫媒病毒检测。针对生产用细胞基质的病毒检测所采取的行动步骤参见表 4(情况B、C 和 E)。
j 如细胞基质/细胞库未检测,应检测。
k 根据产品类型,病毒种子可用于生产疫苗病毒、病毒载体或辅助病毒。病毒种子由已确立的细胞系生成。与基于风险的方法一致,病毒检测应考虑细胞系的来源以及用于制备病毒种子的原材料和试剂,以确保不存在可能来源于细胞基质的外源性病毒,并且确保不存在复制型病毒。应在加工之前对病毒种子进行检测。工作病毒种子(WVS)直接来源于 MVS,因此基于风险评估,针对后续各库进行外源因子检测。另外,不检 MVS,而对每个 WVS 进行全面检测来作为替代手段也是可以的。
1 基于风险评估检测
(+)替代检测阶段
NA=不适用
7.3 病毒清除
应尽可能按照本指导原则的一般原则降低外源性病毒和生产过程中所用病毒的残留物(如辅助病毒和蛋白表达载体)污染的风险。
应采用代表性及经鉴定的缩小规模的系统,验证病毒清除情况。
病毒载体和病毒载体衍生产品的理化特性将决定在产品纯化方案中病毒清除方法的应用。病毒清除验证应包括代表外源、内源以及(如可能)相关辅助病毒的模型病毒。因此第 5 节和第 6 节(包括先验知识的应用)适用,其使用了表 4 所述的特异和非特异模型病毒选择的行动计划。常见的病毒灭活步骤,如用洗涤剂或溶剂/洗涤剂处理,可能适用于与其兼容的产品(如无包膜的病毒载体)。此外,病毒过滤可能更适合于小病毒载体,如 AAV 或基于纳米颗粒的疫苗,其中病毒去除可以基于粒径排阻而实现。适当时,应当进行病毒清除研究,以确定生产工艺相关步骤的病毒下降因子。
示例如下:
- 可以纯化使用杆状病毒/昆虫细胞生产的亚单位蛋白和 VLPs,并且可以通过生产工艺实现高水平的病毒对数下降因子,并通过病毒清除研究进行验证;以及
- 一些病毒载体产品(如 AAV)适用于稳健的病毒清除步骤,确保外源和辅助病毒的清除灭活或去除。
辅助病毒被视为工艺相关病毒污染物。生产工艺需要确保充分清除辅助病毒。基于风险评估的对数下降因子的可接受性。
由于生产过程中的病毒清除工艺可能无法达到与重组蛋白相同的稳健性,这些产品的病毒安全性还依赖于封闭式处理、检测和其他预防性控制措施(见第 2.2、3 和 4 节)。
Hits: 658
Great article on viral safety evaluation! It’s fascinating to see how detailed the guidelines are for ensuring biotech product safety from cell lines. This really highlights the importance of rigorous testing and clearance studies.
Speaking of medication safety and proper usage, I came across an interesting resource that might be relevant to some readers here – it’s about the risks and proper techniques for splitting tablets while traveling. Sorry to drop a link here, but I think it helps illustrate what I’m referring to regarding precise dosing and safety considerations: https://pillintrip.com/tr/article/to-split-or-not-to-split-a-travelers-guide-to-safe-pill-cutting-on-the-go
I’m curious – for biotech products derived from human cell lines, how do regulators typically evaluate the risk of viral contamination when the manufacturing process involves novel excipients or delivery mechanisms that might interact differently with viral clearance steps? Are there any recent updates to the ICH Q5A(R2) guidelines that address emerging viral vectors or advanced therapy medicinal products (ATMPs) more specifically?