
Contents
Assay Development and Validation for Immunogenicity Testing of Therapeutic Protein Products Guidance for Industry
用于检测治疗性蛋白制品的免疫原性的检测法开发和验证行业指南
指南草案
本指南文件仅供征求意见。
关于本草案文件的评论和建议应在联邦公报宣告指南草案可用声明发布后 60 天内提交。关于 本指南的电子评论请提交至 http://www.regulations.gov。关于本指南的书面评论请提交至 Division of Dockets Management(HFA-305),Food and Drug Administration(美国食品药品监 督管理局卷宗管理处),5630 Fishers Lane, rm. 1061, Rockville, MD 20852。所有评论都以登于 联邦公报上的可用声明卷宗号作为识别标记。
关于本草案文件的问题,请联系(CDER)Susan Kirshner ,电话 301-827-1731;(CBER)对 外信息、外联和发展办公室,800-835-4709 或 240-402-8010;或(CDRH)交流教育办公室, 800-638-2041 或 301-796-7100。
美国卫生和公众服务部
美国食品药品监督管理局
药品评价和研究中心(CDER)
生物制品评价和研究中心(CBER)
医疗器械和放射健康中心(CDRH)
2016 年 4 月药品质量/CMC
修订 1
用于检测治疗性蛋白制品的免疫原性的检测法开发和验证行业指南
如需副本,请联系:
药品信息处对外信息办公室
药品评价和研究中心
美国食品药品监督管理局
10001 New Hampshire Ave., Hillandale Bldg., 4th Floor
Silver Spring, MD 20993-0002
电话:855-543-3784 或301-796-3400;传真:301-431-6353
电子邮件:druginfo@fda.hhs.gov
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
和/或
对外信息、外联和发展办公室
生物制品评价和研究中心
美国食品药品监督管理局
10903 New Hampshire Ave., Bldg. 71, Room 3128
Silver Spring, MD 20993-0002
电话:800-835-4709 或240-402-8010
电子邮件:ocod@fda.hhs.gov
和/或
交流教育办公室
CDRH-工业和消费者教育处
医疗器械和放射健康中心
美国食品药品监督管理局
10903 New Hampshire Ave., Bldg. 66, Room 4621
Silver Spring, MD 20993-0002
电话:800-638-2041 或301-796-7100;传真:301-847-8149
电子邮件:DICE@fda.hhs.gov
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/default.htm
美国卫生和公众服务部
美国食品药品监督管理局
药品评价和研究中心(CDER)
生物制品评价和研究中心(CBER)
医疗器械和放射健康中心(CDRH)
2016 年 4 月药品质量/CMC
|
一旦定稿后,本草案指南代表美国食品药品监督管理局(FDA)目前关于该问题的想法。它对任何人 都不赋予任何权利,也不对 FDA 或公众具有约束性。您可以采用其他方法,只要该方法符合所适用 法规和条例的要求。要讨论替代方法,请联系如标题页所列的负责本指南的 FDA 工作人员。 | |
包含非约束性建议
草案–不实施
I. 引言
本指南提供了建议,以促进行业开发和验证用于评估治疗性蛋白制品在临床试验中的免疫原性的免疫 检测法。具体来说,本文件包括关于筛选检测法、确证性检测法、滴定检测法和中和检测法的开发和 验证的指导。2,3 鉴于本指南的目的,免疫原性定义为治疗性蛋白制品对自身及相关蛋白产生免疫反应 或诱导免疫相关不良临床事件的倾向性。本文件中提供的用于检测法开发和验证的建议适用于检测抗 药物抗体(AD)的检测法。4 本指南也可能适用于某些复方产品,具体取决于具体情况。5
1 本指南是由美国食品药品监督管理局药物评价和研究中心(CDER)医疗政策办公室联合医疗器械和放射健康中心编制的。
2 本文件没有具体讨论用于动物研究的抗药物抗体(AD)检测法的开发或验证; 然而,所讨论的一些概念与非临床试验的 ADA 研究的设计相关。关于动物毒理学研究中的免疫原性评估的更多信息,参见国际协调会议(ICH)行业指南 S6(R1) 生物技术药物临床前安全性评价。另见治疗性蛋白制品免疫原性评估行业指南,其中更详细地讨论了“动物研究效用”主题。 我 们 定 期 更 新 指 南 。 为 了 确 保 您 拥 有 最 新 版 本 的 指 南 , 检 查 FDA 指 南 网 页 http://www.fda.gov/RegulatoryInformation/Guidances/default.htm。
3 关于拟定生物仿制药生物制品的临床免疫原性评估信息,参见行业指南证明与参照药具有生物相似性的科学考虑要点。
4 本指南不涉及用于评估对传染性疾病适应症的预防性和治疗性疫苗的免疫应答的免疫原性检测法。
5 关于复方产品的一般信息,请参见http://www.fda.gov/CombinationProducts/default.htm。
本文件不讨论可能造成免疫原性的产品和患者风险因素。6 本指南,包括本指南中使用的术语的任何 讨论,不适用于体外诊断产品。7 本指南修订了 2009 年 12 月发布的草案行业指南用于治疗性蛋白免 疫原性检测的检测法开发。为了清楚起见,本指南中的信息已重新组织,包括关于滴定检测法和确证 性检测法的新信息。
一般来说,FDA 的指南文件不构成有强制力的法律责任。相反,指南描述的是 FDA 当前关于某问题 的想法,仅作为建议,除非引用特定的法规或法令要求。FDA 指南中出现的“应(当)”的词义是指建 议或推荐,而非要求。
II. 背景
患者对治疗性蛋白制品的免疫应答具有影响产品安全性和有效性的潜力。8 患者免疫应答的临床效果 是高度可变的,从对患者健康完全没有影响到极其有害的影响。ADA 形成的检测和分析是了解潜在 的患者免疫应答应的有用工具。在临床试验期间观察到的关于免疫应答的信息,特别是 ADA 诱导的 发生率和 ADA 应答对治疗性蛋白制品安全性和有效性的影响,对于任何治疗性蛋白制品开发程序是 至关重要的。因此,如果适用,这些信息应作为题为“免疫原性”的“不良反应”章节的一部分包括在处 方信息中。因此,开发用于测量 ADA 应答的有效、灵敏且具有专属性和选择性的检测法是治疗性蛋 白制品开发的关键方面。
III. 一般原则
患者对治疗性蛋白质制品产生免疫应答的风险将随产品变化而变化。
6 参见治疗性蛋白制品免疫原性评估行业指南,其中更详细地讨论这些主题。
7 根据 21 CFR 809.3(a),“体外诊断产品是旨在用于诊断疾病或其他病症(包括确定健康状况)的试剂、仪器和系统,以治 愈、减轻、治疗或预防疾病或其后遗症。此类产品旨在用于收集、制备和检查从人体采集的样本。这些产品是“美国联邦食 品、药品和化妆品法案”(法案)第 201(h)节中定义的医疗器械,也可能是符合“公共卫生服务法案”第 351 节的生物制 品。
8 参见治疗性蛋白制品免疫原性评估行业指南。
FDA 建议采用基于风险的方法来评价和减轻与影响治疗性蛋白制品安全性和有效性效的治疗性蛋白 制品相关的免疫应答或与免疫相关的不良临床事件。9 免疫应答可能具有多种效应,包括中和活性和 诱导超敏反应的能力。免疫原性检测应设计为检测可介导不期望的生物或生理结果的 ADA。
筛选检测法(也称为结合抗体(BAb)检测法)用于检测与治疗性蛋白制品结合的所有抗体。使用确 证性检测法建立 BAb 对治疗性蛋白制品的特异性。使用滴定和中和检测法进一步表征 ADA 。使用滴 定检测法表征 ADA 应答的量级。重要的是使用滴定检测法来表征这种量级,因为 ADA 对安全性和 有效性的影响可能与 ADA 滴度和持续性而不是发生率相关(Cohen and Rivera 2010)。中和检测法评 估 ADA 干扰治疗性蛋白制品-靶标相互作用的能力。因此,中和抗体(NAb)是 BAb 的亚类。重要 的是使用滴定检测法来表征 ADA 的中和活性,因为 ADA 对安全性和有效性的影响可能与 Nab 活性 而不是 ADA 发生率相关(Calabresi, Giovannoni, et al.2007; Goodin, Frohman, et al.2007; Cohen and Rivera 2010)。类似地,在一些情况下可能重要的是确定 NAb 滴度。额外表征检测法(例如分型、表 位作图和评估例如与内源配对物或其他产品的交叉反应性)可能是有用的。
在治疗性蛋白制品开发期间设计、开发和验证 ADA 检测法的最佳时间取决于产品的风险评估 (Mire-Sluis, Barrett, et al.2004; Gupta, Indelicato, et al.2007; Shankar, Devanarayan, et al.2008; Gupta, Devanarayan, et al.2011)。申办者应提供免疫原性检测范例的理论依据,最好在研究新药申请(IND) 阶段,I 期研究期间。因为当免疫原性造成高临床风险(例如,评估具有非冗余内源配对物的治疗性 蛋白产品)和需要关于患者应答的实时数据时,ADA 检测法是关键的,所以申办者应当在 I 期研究之 前和期间尽早实施初步验证的检测法, 并实时获得数据。实时评估需要在采样后、样品建库前和给 药方案允许时额外给药前尽快对样品进行分析。在较低风险的情况下,申办者可以对患者样品进行建 库,以便在合适的检测法可用时对样品进行检测。FDA 鼓励申办者在 I 期和 II 期研究期间使用合适的 检测法检测样品。应使用经完全验证的检测法对来自关键性研究的样品进行检测。在许可申请时,申 办者应提供支持该检测法的全面验证的数据。关于 ADA 样品采集时间的建议见第 VII. A 节。10
9 参见治疗性蛋白制品免疫原性评估行业指南。
10 参见治疗性蛋白制品免疫原性评估行业指南,其中更详细地讨论免疫原性风险评估和减轻考虑因素。关于用于免疫原性 检测的适当检测法的开发和验证的指南也可以在 ICH 行业指南 Q2A 分析方法验证文本和 Q2B 方法验证:方法获得。
用于检测 ADA 的检测法有助于理解治疗性蛋白制品的免疫原性、安全性和有效性。然而,ADA 的检 测依赖于检测法的关键操作参数(例如,灵敏度、专属性),不同检测法之间不同。11 尽管关于 ADA  发生率的信息通常包括在不良反应章节的“免疫原性”子章节的处方信息中,但 FDA 警告说,即使对 于具有序列或结构同源性的产品,ADA 发生率的比较也可能是误导性的。这是因为 ADA 形成的检测 高度依赖于检测法的灵敏度和专属性。另外,在检测法中观察到的 ADA(包括 NAb)阳性发生率可 能受到诸如方法、样品处理、样品采集时间、伴随药物和疾病状况等因素的影响。因此,即使使用经 过完全验证的检测法,比较具有相同适应症的结构同源性治疗性蛋白制品的免疫原性比率是不牢靠的。 当需要具有同源性的不同治疗性蛋白制品之间的免疫原性的真实比较时,应当通过使用标准化检测法 在对于两种治疗性蛋白制品具有相等的灵敏度和专属性的相同条件下进行头对头临床研究来获得。12
本指南中提供的关于检测法开发和验证的建议是基于 FDA 在审查免疫原性递交资料时遇到的常见问 题。申办者应联系 FDA 以获得任何产品特定的指导。应当与 FDA 讨论 分型和交叉反应性检测法设 计。也可以咨询其他出版物以获得更多见解(参见 Mire-Sluis, Barrett, et al.2004; Gupta, Indelicato, et al.2007; Shankar, Devanarayan, et al.2008; Gupta, Devanarayan, et al.2011)。一般来说,FDA 建议申办者 开发针对灵敏度、专属性、选择性、精确度、重复性和稳定性进行优化的检测法(参见第 IV.C 节至 第 G 节)。
IV. 检测法设计元素
本节适用于用于检测 ADA 的所有类型的检测法,除非另有说明。
A. 检测策略
1. 多层检测方法
FDA 推荐多层 ADA 检测方法,因为一些临床试验的规模和在几个时间点检测患者样品的必要性。在 该范例中,最初使用快速、灵敏的筛选检测法来评估临床样品。初始筛选检测法应对低水平的低亲和 力和高亲和力 ADA 敏感(参见第 V.A 节)。然后对在筛选检测法检测中呈阳性的样品进行确证性检测 法检测以证明 ADA 对治疗性蛋白制品具有特异性。
11 关于多种检测法类型的更广泛讨论,参见美国药典(USP)通则 1106 免疫原性检测法-用于检测抗药物抗体的免疫检测法 的设计和验证。
12 关于拟定生物仿制药产品的信息,参见行业指南证明与参照药具有生物相似性的科学考虑要点。
例如,竞争检测法可以确认抗体与治疗性蛋白制品特异性结合,并且筛选检测法中的阳性结果不是试 验血清或检测试剂与检测法环境中的其他材料(例如塑料或其他蛋白)的非特异性相互作用的结果。
在确证性检测法中鉴定为阳性的样品应在其他检测法(如滴定和中和检测法)中进行进一步表征。在 一些情况下,可能需要检测与具有同源性的其他蛋白(例如相应的内源性蛋白)的交叉反应性的检测 法。此外,一旦含有抗体的样品经确认为阳性,也可推荐用于评估抗体的同种型及其表位特异性的检 测。
2. 免疫球蛋白同种型
初始筛选检测法应能够检测所有相关的免疫球蛋白(Ig)同种型。对于非粘膜给药途径,并且在不存 在过敏反应的情况下,预期的ADA 同种型是IgM 和IgG。对于粘膜给药途径,也预期IgA 同种型ADA 。 虽然 FDA 预期在筛选检测法中检测到所有相关同种型,但筛选检测法不必确定检测到哪些同种型。 例如,使用桥接形式的检测法可能不提供关于正在检测哪些同种型的信息。桥接检测法形式理论上可 以检测大多数同种型的抗体,但可能不检测 IgG4 同种型。在一些情况下,申办者应当开发区分抗体 同种型的检测法。例如,对于涉及过敏性风险的治疗性蛋白制品,应该开发抗原特异性 IgE 检测法。 此外,IgG4 抗体的产生与在长期抗原暴露的条件下产生的免疫应答有关,例如采用因子 VIII 治疗, 以及在接受红细胞生成素治疗的纯红细胞发育不全患者中(Matsumoto, Shima, et al.2001; Aalberse and Schuurman 2002)。因此,根据临床问题,可能需要评估特定同种型。
3. 表位特异性
FDA 建议申办者对整个治疗性蛋白制品和(如果相关)其内源配对物进行初步筛选检测。对于一些治 疗性蛋白制品,申办者可能需要研究特异性表位(特异性产生免疫应答)的 ADA 。例如,建议测定 一些融合分子的表位特异性,因为两个分子连接的区域可能形成新抗原,并且可能产生对该区域的免 疫应答。由于表位扩散,可能发生对分子其他部分的免疫应答,导致产生治疗性蛋白制品或其内源性 配对物抗体(Prummer 1997; Miller, Korn, et al.1999; Disis, Goodell, et al.2004; Thrasyvoulides, Liakata, et al.2007; van der Woude, Rantapaa-Dahlqvist, et al.2010; Hintermann, Holdener, et al.2011)。对于这些治疗 性蛋白制品,FDA 鼓励申办者研究免疫级联中的起始事件。这方面的知识可允许修饰蛋白以降低其潜 在的免疫原性。类似地,对于经过修饰(例如聚乙二醇化修饰)的治疗性蛋白制品,申办者应该开发 检测法以确定 ADA 对蛋白组分的特异性以及对治疗性蛋白制品的修饰。
另见第 IV.K.4 节和第 IV.K.5 节。
B. 检测法切点
检测法的切点是将样品响应定义为阳性或阴性的检测法的响应水平。在第 V 节和第 VI 节中提供了确 定响应检测法类型的切点所特有的信息。确定适当的切点对于确保可接受的检测法灵敏度至关重要。
检测法的切点可以受到无数干扰因素的影响,例如预先存在的抗体、类风湿因子(RF)、人抗小鼠抗 体以及基质中产物相关材料或同源蛋白的水平。当定义切点时,应当在检测法开发的早期考虑这些因 素。因为来自不同目标人群和疾病状态的样品可能具有可导致检测法的背景信号变化的组分,因此对 于所研究的离散人群可能需要不同的切点。
切点应使用来自初治受试者的样品统计学确定。13 通过使用这些样品进行重复检测运行,可以估计检 测法的变异性。在检测法开发期间,可以使用少量样品来估计切点。这可能使用来自初治受试者的少 至 5-10 份样品进行。
用于确定切点的具体方法将取决于各种因素。具体来说,因为切点应当鉴别产生超过检测法的变异性 的信号的任何样品,所以申办者应当在确定切点时考虑统计学确定的异常值以及真阳性样品的影响。 申办者应提供删除任何数据点的理由,以及用于确定其作为异常值的状态的相应方法。阳性值和样品 可能衍生自非特异性血清因子或患者样品中存在的预先存在的抗体(Ross, Hansen, et al.1990; Turano, Balsari, et al.1992; Coutinho, Kazatchkine, et al.1995; Caruso and Turano 1997; van der Meide and Schellekens 1997; Boes 2000)。尽管在健康个体中存在针对多种内源性蛋白的预先存在的抗体,但是在 一些疾病状态中这些抗体可以高得多。申办者应该鉴别存在预先存在的抗体的样品,例如通过免疫耗 竭方法,并从切点分析中去除它们。如果预先存在的抗体的存在是混杂因素,则可能有必要基于暴露 之前和之后的个体患者结果之间的差异指定阳性应答或切点。通过仔细的设计考虑,例如利用样品的 最低稀释倍数(MRD),去除分析中的统计异常值,最小化干扰因子的影响,改善检测法药物耐受性, 并使用考虑预先存在的抗体的方法,可能获得合理的值来定义检测法切点。
13 初治受试者可以是健康个体或未暴露于治疗性蛋白制品的患者人群,这取决于检测法开发或验证的阶段以及样品的可用 性。申办者应提供所使用样品是否合适的证明。
C. 灵敏度
1. 检测法灵敏度
申办者应当确定检测法的灵敏度能够确保报告免疫原性率时具有充分的置信度。检测法灵敏度表示抗 体制剂始终产生等于针对该特定检测法确定的切点的阳性结果或读出的最低浓度。14 FDA 建议筛选 和确证性 ADA 检测法达到至少 100 毫微克/毫升(ng / mL)的灵敏度。虽然传统上 FDA 已经建议至 少250-500 ng/mL 的灵敏度,最近的数据表明低至 100 ng/mL 的浓度可能与临床事件相关(Plotkin 2010; Zhou, Hoofring, et al.2013)。然而,应当理解,中和检测法可能不总是达到该灵敏度水平。
检测法应当具有足够的灵敏度以能够在 ADA 的量达到可以与改变的药代动力学、药效学、安全性或 有效性特征相关的水平之前检测低水平的 ADA 。因为将在生物基质的存在下评估患者的抗体水平,
所以应当使用相同生物基质(例如,具有与稀释剂相同的抗凝血剂的血清或血浆,来自目标人群)的 相关稀释来进行检测法灵敏度的检测)。最终的灵敏度应该表示为可检测的抗体的质量/未稀释基质mL。 因此,应在考虑到 MRD 后报告检测法灵敏度。检测法灵敏度不应报告为滴度。在开发期间,应使用 自初治受试者的单个以及合并样品来评估灵敏度,以便可以确定阴性对照的适用性。
应通过检测已知浓度的阳性对照抗体在合并的阴性对照基质中的连续稀释来确定检测法灵敏度。稀释 系列应不大于 2 或 3 倍,并且应检测至少5 个稀释度。或者,可以通过将稀释曲线的线性部分内插至 检测法切点来计算灵敏度。如前所述,检测法灵敏度应以质量单位/未稀释基质体积的报告。
应使用治疗性蛋白制品特异性抗体的纯化制剂来确定检测法的灵敏度,使得检测法灵敏度可以以质量 单位/ mL 基质报告。用于评估灵敏度的抗体可以采取亲和纯化多克隆制剂或单克隆抗体(mAb)形式。
应使用含有略高于检测法灵敏度的 ADA 浓度的低阳性系统适用性对照,以确保检测法的灵敏度在各 检测运行中一致。
14 关于相对灵敏度的讨论,参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗体的免疫检测法的设计和验证。
低阳性系统适用性对照应设计为在 1%的运行中失败(参见第IV.I.1 节)。
2. 药物耐受性
治疗性蛋白制品或存在于血清中的内源配对物可能干扰检测法的灵敏度。具体来说,如果产品相关材 料存在于试验样品中,则 ADA 和治疗性蛋白制品之间可以形成复合物(也称为 ADA-药物复合物), 其阻止在检测形式中检测 ADA 。这是因为 ADA 检测法通常设计用于检测未复合的 ADA 。在预期的 干扰性治疗性蛋白制品水平存在下的检测法灵敏度的评估,也称为检测法的药物耐受性,对于理解方 法用于检测给药患者中 ADA 的适用性是至关重要的。15 FDA 建议申办者在检测法开发早期检查检测 法药物耐受性。申办者可以通过在不存在或存在不同量的所考虑的治疗性蛋白制品的情况下故意地将 不同的已知量的纯化 ADA 添加到单独的 ADA 阴性对照样品中并定量测定治疗性蛋白制品是否干扰 ADA 检测来检查药物耐受性。应该比较在不存在和存在不同量的所考虑的治疗性蛋白制品时获得的 结果。在指定程度的抑制所需的抗体量和治疗性蛋白制品量之间应当存在相关性。来自药代动力学研 究的数据可用于确定最佳的样品采集时间。酸解离预处理或其他方法可用于破坏循环 ADA 药物复合 物,这可导致检测法药物耐受性增加。如果申办者在治疗性蛋白制品已经衰减至其不干扰检测法结果 的水平时采集患者样品,则治疗性蛋白制品的干扰可以最小化。
D. 专属性和选择性
证明检测法专属性和选择性对于免疫原性检测结果的解释是关键的。专属性是指一种方法检测结合治 疗性蛋白制品但不结合检测法组分(如表面或试剂)的 ADA 的能力。检测法应该专门检测目标分析 物,在这种情况下是 ADA 。16 ADA 检测法的选择性是其在可能含有潜在干扰物质的基质(如血清或 血浆)中鉴别治疗性蛋白制品特异性 ADA 的能力。检测法结果可能受到来自基质或来自在机治疗性 蛋白制品的干扰的影响。17 缺乏检测法专属性或选择性可导致假阳性结果,这可能模糊 ADA 应答与 临床安全性和有效性测量指标之间的关系。
15 参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗体的免疫检测法的设计和验证。
16 参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗体的免疫检测法的设计和验证。
17 参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗体的免疫检测法的设计和验证。
证明对 mAb、Fc-融合蛋白和 Ig-融合蛋白的抗体应答的专属性和选择性构成特殊的挑战,因为人血清 中的高浓度 Ig 。申办者应明确证明检测方法专属性地检测抗 mAb,而不是 mAb 产品本身、非特异性 内源性抗体或检测中使用的抗体试剂。类似地,对于具有高 RF 发生率的患者人群,申办者应该证明 RF 不干扰检测方法。宿主细胞蛋白和其他产物相关杂质也可能干扰证明检测法专属性和选择性。
解决专属性和选择性的直接方法是证明结合可以被可溶性或未标记的纯化治疗性蛋白制品阻断。一种 方法是将阳性和阴性对照抗体样品与所考虑的纯化治疗性蛋白制品或其组分一起孵育。在相关治疗性 蛋白制品或其组分存在下的信号抑制证明该应答具有专属性和选择性。对于 mAb 产品的应答,纳入 具有相同 Fc 但不同可变区的另一 mAb 可能是关键的。对于对其他蛋白的应答,可以使用相似大小和 电荷的非相关蛋白。如果检测法对于所讨论的蛋白具有专属性和选择性,则通常在溶液中添加该蛋白 应当减少对背景或切点的反应,而添加相似大小和电荷的非相关蛋白应该没有效果。相反,所讨论的 蛋白的添加应对非相关蛋白特异性抗体几乎没有影响。应通过进行回收率研究进一步评价选择性,其 中将阳性对照抗体加标到限定浓度的基质中,并将阳性对照抗体信号与从加标到检测缓冲液中的抗体 获得的信号进行比较。
1. 基质干扰
一个重要的考虑因素是由样品和稀释剂组成的检测基质的干扰如何影响检测法性能。基质中除治疗性 蛋白制品之外的组分可能干扰检测法结果。例如,在样品采集期间使用的不同抗凝剂可能在检测法中 具有不同的效果,潜在地影响检测法灵敏度和线性。申办者应评估不同的盐抗凝血样品采集溶液对检 测法结果的影响。
血清或血浆中的内源和外源组分可能影响检测法结果,并且通常需要稀释患者样品用于检测以使这种 影响最小化。申办者应通过进行加标-回收率研究来检查这些干扰物的影响。申办者应定义用于在进 行验证研究之前用于制备患者样品的稀释因子,评估该基质对检测法结果的潜在干扰(关于 MRD , 参见第 IV.D.2 节)。
与治疗性蛋白制品化学相关的缓冲液组分也可能干扰检测。例如,聚山梨醇酯化学上类似于聚乙二醇 (PEG),因此可能干扰抗 PEG 抗体的检测。在检测法开发过程中应仔细考虑缓冲液的化学组成。
申办者可以通过在不存在或存在不同的基质组分的情况下将不同的已知量的纯化 ADA 加标到检测缓 冲液中来检查基质干扰。比较单独的缓冲液中的 ADA 与基质中的 ADA 的回收率可以提供来自基质 组分的干扰程度的输入。此外,这种分析可以指导针对样品检测推荐的 MRD 的决定。此外,申办者 应该检查影响患者样品的其他参数,例如溶血、脂血症、胆红素的存在,以及患者人群可能使用的伴 随药物的存在。具有非常高的抗体滴度的样品可能需要另外的检测,例如在确证性检测中使用不同稀 释度的竞争产品,以确保它们的鉴别。
2. 最低稀释倍数
如果未稀释,则基质组分可导致非特异性信号,从而模糊阳性结果。因此,经常需要稀释患者样品以 保持检测 ADA(敏感性)的合理能力。理想地,MRD 是产生接近检测稀释剂的信号的信号并且允许 最高的信噪比的样品稀释度。MRD 通常为 1:5 至 1:100。
FDA 建议申办者利用来自初治受试者的一组适当数量的样品确定 MRD。MRD 的测定通常包括连续稀 释初治 ADA 阴性样品,以及与缓冲液中相同量的抗体相比,检测连续稀释基质中的已知量的纯化抗 体(高、中和低浓度)。这确保了在整个检测法范围内的合理的信噪比。MRD 应使用至少 10 个单个 血清样品计算;适当数量的样品将取决于各种因素,包括单份样品的变异性。
虽然申办者最终选择的 MRD 将取决于检测法设计和患者人群,但 FDA 建议稀释度不超过 1:100 。当 患者实际上可能具有低水平的治疗性蛋白制品特异性抗体时,更高的稀释度可能导致假阴性应答的鉴 定,这种情况的发生可能与显着改变的药代动力学、药效学, 安全性或有效性特征相关。然而,在一 些情况下,可能需要更大的初始稀释度,并且应考虑这种稀释度对检测法灵敏度和免疫原性风险评估 的总体影响。
E. 精密度
精密度是对方法中相同材料运行的一系列测量的变异性的度量。结果应在检测运行中和检测运行之间 重现,以确保足够的精密度。18 证明检测法精密度对于 ADA 的评估至关重要,因为检测法变异性是 确定切点和确保低阳性样品被检测为阳性的基础。
18 关于精密度的更多信息,参见生物分析方法验证行业指南。另外参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗 体的免疫检测法的设计和验证。
为了提供可靠的估计,申办者应当评价检测法反应的检测内(重复性)和检测间(中间精密度)变异 性。
F. 重现性
如果在研究期间由两个或更多个独立实验室进行检测,重复性是重要的考虑因素,并且申办者应确定 每个实验室产生的数据的可比性。19 此外,在所确定的检测法操作条件下(例如,使用相同的仪器平 台),检测法应在不同实验室之间具有相同的精密度。
G. 耐用性和样品稳定性
检测法耐用性是检测法在正常使用期间的可靠性的指示 20,并且通过在常规实验室实践中检测法保持 不受在相关的真实情况下预期的方法和仪器性能的较小的但有意的变化影响的能力评估。例如,温度、 孵育时间或缓冲液特征(例如 pH 值和盐浓度)的变化都可影响检测法结果。生物检测法的复杂性使 得它们对检测条件的变化特别敏感,并且评价和优化参数(例如细胞通道数、孵育时间和培养基组分) 是必要的。申办者应在开发阶段检查耐用性,如果检测法中特定步骤的微小变化影响结果,则应采取 特定预防措施来控制其变异性。FDA 建议以检测时保留抗体反应性的方式储存患者样品。FDA 建议 申办者避免冻融循环,因为冻融患者样品也可能影响检测法结果。然而,评估阳性对照抗体的长期稳 定性的研究可能是有用的。21
H. 格式选择
许多不同的检测法格式和仪器可用于检测 ADA 。这些包括但不限于直接结合检测法、桥接检测法和 平衡结合检测法。每种检测法格式具有优点和缺点,包括通量的快速性、灵敏度、选择性、动态范围、 检测各种 Ig 同种型的能力、检测快速解离抗体的能力和试剂的可用性。这些检测法格式中的每一种 之间的主要差异之一是清洗次数和活力,这可以对检测法灵敏度产生影响。
19 关于重现性的更多信息,参见生物分析方法验证行业指南。另外参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗 体的免疫检测法的设计和验证、USP 通则 1225 药典方法验证以及 ICH 行业指南分析法方法验证:方法。
20 关于耐用性的更多信息,参见 ICH 行业指南 Q2B 分析方法验证:方法。另外参见 USP 通则 1106 免疫原性检测法-用于检 测抗药物抗体的免疫检测法的设计和验证。
21 关于稳定性研究的更多信息,参见生物分析方法验证行业指南。
应评价所有检测法检测在早期免疫应答中是常见的快速解离抗体(例如IgM)的能力。未能检测在对 治疗性蛋白制品的早期免疫应答中这样的抗体可能导致真阳性抗体样品的低检测率。表位暴露也是重 要的,因为与塑料结合或与其他试剂(例如报道分子(即荧光染料、酶或生物素))偶联可导致抗原 的构象变化,这可掩盖、暴露、修饰或破坏治疗性蛋白制品上的相关抗体结合位点。
I. 试剂选择
用于 ADA 检测的检测法的许多组分可以是标准的或从商业来源获得,例如,市售试剂,例如在确证 性检测法的耗竭方法中使用的蛋白 A/G 包被树脂。然而,特定检测法可能需要其他特定组分,包括阳 性对照抗体、阴性对照和系统适用性对照。
1. 阳性对照抗体的开发
申办者可以使用不同或相同的阳性对照抗体来建立和监测在检测法性能的常规评估期间的系统适用 性,以及确定所采用的检测法是否适合目的。对于系统适用性对照,在调整至控制切点和动态范围水 平的浓度下使用的阳性对照抗体(单克隆或多克隆)可能是合适的。
阳性对照抗体通常通过由免疫动物在不存在或存在佐剂的情况下产生。FDA 建议使用治疗性蛋白制品 对由免疫动物产生的阳性对照抗体进行亲和纯化。这种方法丰富了 ADA 的多克隆抗体制剂,这使得 能够更准确地解释灵敏度评估结果。应仔细考虑产生阳性对照抗体时动物种属的选择。例如,如果抗 人 Ig 试剂将用作检测患者抗体的第二试剂,则阳性对照抗体和质量控制(QC)样品应当可通过相同 的试剂检测。当阳性对照抗体不能通过相同的试剂检测时,可能需要另外的第二试剂来检测阳性对照 抗体。在这些情况下,应该实施用于检测人抗体的第二试剂的另外的阳性对照抗体,以确保试剂按照 预期进行。在一些情况下,申办者能够利用患者样品产生阳性对照抗体。22 尽管这样的抗体可以是非 常有价值的,但是这样的样品通常在早期试验中不可用。或者,单个 mAb 或 mAb 组可用于阳性对照 抗体。申办者应与 FDA 讨论在申办者不能产生阳性对照抗体的罕见情况下检测法开发和验证的替代 方法。
22 需要患者的正确知情同意,并应提前计划。
理想地,用于确定检测法对于相应检测目的的适用性的阳性对照抗体应反映将在人中发生的预期免疫 应答。对于治疗性 mAb,申办者应特别考虑用于检测法的阳性对照抗体的选择。当用嵌合、人源化或 人 mAb 免疫动物以产生阳性对照抗体时,体液应答可针对人 Fc 而不是分子的可变区。这样的阳性对 照抗体可能与其中应答主要针对抗原结合区的患者中的预期免疫应答不相关。
一旦已鉴定阳性对照抗体的来源,则申办者应使用该来源来评估检测法性能特征,例如灵敏度、选择 性、专属性和重现性。FDA 建议申办者生成和保留阳性对照抗体溶液用作质量或系统适用性控制。对 于检测法开发和验证,稀释应当代表检测法中的高、中和低值。甚至对于定性检测法来说,甚至需要 了解检测法性能在广泛的抗体浓度范围内是否可接受。虽然应该使用高值和低值 QC 样品,但是通常 不需要用于检测 ADA 的中值 QC 样品来监测在检测法性能的常规评估期间的系统适用性。
2. 阴性对照的开发
对于阴性对照样品,建议在可能的情况下,对照人群应具有相同的疾病状况。对照样品应代表相似的 性别、年龄和伴随药物,以使样品基质代表研究人群。类似地,应当以与研究样品相同的方式收集和 处理对照样品,例如关于使用的抗凝血剂的类型、样品体积以及样品制备和储存,因为这些预分析变 量可以影响检测法中对照样品的性能。通常情况下,这种对照样品在开发或研究前验证活动期间不可 用。在这些情况下,使用购买的样品或来自健康供体的样品是可接受的,但是当来自适当目标人群的 初治受试者的样品变得可用时,应当确认检测法性能的重要参数,例如切点、灵敏度和选择性。
FDA 建议申办者建立验证研究和患者样品检测的阴性对照。在这方面,来自适当数量的初治受试者的 血清池可以用作有用的阴性对照。重要的是,阴性对照获得的值应当低于但接近在检测法中针对所检 测的患者人群所确定的切点。产生远低于来自用于建立切点的单个血清样品的平均值的值的阴性对照 可能不能用于确保适当的检测法性能。
3. 检测试剂考虑因素
合适的检测试剂(即报道分子)的选择取决于所选择的检测法格式。关键是最小化来自检测试剂的非 特异性信号。
所选择的检测试剂应具有特定检测法所需的足够的灵敏度。在决定检测试剂时应考虑这些因素。
4. 非特异性结合控制
每种试剂,从微量滴定板的塑料到显影剂,可以影响检测法灵敏度和非特异性结合。最关键的因素之 一是选择适当的检测缓冲液和用于防止非特异性结合固体表面的封闭试剂。申办者应仔细考虑清洗步 骤的数量和时间以及添加到检测缓冲液(即封闭或清洗缓冲液)中的洗涤剂,以减少背景噪声但仍保 持灵敏度。多种蛋白质可以用作封闭试剂以提供可接受的信噪比。然而,这些蛋白质可能不能全部在 特异性免疫检测法中等效运行。例如,它们可能不能很好地结合所有类型的固相或可能显示出与检测 试剂的非预期交叉反应性。因此,申办者可能需要检测几种阻断剂以优化检测法性能。此外,包括未 包被的孔不足以评估非特异性结合。相反,确定 ADA 结合可能存在于样品中的相似大小和电荷的不 相关蛋白的能力可能证明是结合特异性的更好检测。
J. 定性和半定量检测法的报告结果
可以使用几种方法来报告阳性抗体反应,并且应当根据具体情况评价所使用方法的适用性。最常见的 方法是定性方法,患者报告为具有阳性或阴性抗体应答。
对于经确认为 ADA 阳性的患者,测定抗体水平可以提供信息,因为这允许 ADA 水平及其对安全性 和有效性的影响的分层评估。除非确定 ADA 水平,否则这些关系可能不清楚。当适当时,阳性抗体 应答可以报告为滴度(例如,给出检测法切点或正好高于检测法切点的读数的最高稀释度的倒数)。 MRD 应该在滴度的计算中考虑,并且在报告滴度时提供。以滴度单位报告的抗体水平是适当的,并 且医学界通常理解。值也可能报告为每体积血清中和的治疗性蛋白制品的质量单位的量,需要提醒的 是,这些是任意的体外检测法单位,并且不能用于直接评估体内治疗性蛋白制品的可用性。
除非所使用的检测法允许独立测定质量,以质量单位报告的抗体水平通常是不可接受的,因为它们基 于来自用阳性对照抗体产生的标准曲线的数据的插值,并且不能假定参比标准品和供试品之间的平行 性。因此,FDA 不认为申办者以质量单位报告患者抗体结果不必要也不可取,除非(1)结果通过定 量方法确定,或(2)普遍接受和可获得来源的经验证抗体可用作对照,并且已经证明了对照抗体和 患者样品的稀释曲线之间的平行性。此外,即使证明了平行性,因为参比标准品和供试品可能含有不 同的抗体群,无法计算绝对质量单位。
因此,FDA 认为报告的质量单位是相对值而不是绝对值。
K. 检测法开发的其他考虑因素
许多因素可以影响抗体水平的评估,例如患者样品变异性、在基于细胞的中和生物检测法用于产生中 标准曲线的细胞的治疗性蛋白制品-剂量反应、ADA 的亲和力和亲合力以及确证性检测法中竞争产品 的浓度。考虑这些因素对于理解和分析检测法变异性以及避免错误是重要的。应考虑的常见因素包括:
1. 预先存在的抗体
医学文献中越来越多的证据表明对许多自身蛋白具有特异性的 B 细胞和 T 细胞是天然存在的,并且甚 至可能在一些疾病状态中升高,例如在进行细胞因子治疗或患有多种免疫性或免疫炎性疾病的患者中 (Coutinho, Kazatchkine, et al.1995; van der Meide and Schellekens 1997; Boes 2000)。例如,干扰素抗体 可以在正常个体中发现(Ross, Hansen, et al.1990; Turano, Balsari, et al.1992; Caruso and Turano 1997)。 令人不会感到惊讶的是,受试者可能具有针对外来抗原(例如细菌产物)的预先存在的抗体,这最有 可能是暴露于微生物或交叉反应性的结果。预先存在的抗体可能具有临床疗效,并且可能影响所检测 的治疗性蛋白制品的有效性。当存在预先存在的抗体时,可能需要定性筛选检测法的替代方法来评估 ADA 的数量和质量。例如,使用半定量检测法类型(例如滴定检测法)(参见章节 V.C 和VI.D)检测 样品中 ADA 增加可提供关于治疗性蛋白制品对产品免疫原性的影响的信息,而定性检测法不提供这 一信息。
2. 类风湿因子
当 RF 存在于血清或血浆中时,测量对具有 Ig 尾部的治疗性蛋白制品(例如 mAb 和 Fc-融合蛋白)的 免疫应答可能是特别困难的。RF 通常是识别 IgG 的 IgM 抗体,尽管已经注意到其他 Ig 特异性。因此, RF 将结合 Fc 区,使得看起来存在治疗性蛋白制品的特异性抗体。已经证明用于使来自 RF 的干扰最 小化的几种方法是有用的,包括用阿斯巴甜处理(Ramsland, Movafagh, et al.1999)并仔细优化试剂浓 度以减少背景结合。当在存在 RF 的临床环境中检查对 Fc 融合蛋白的免疫应答时,FDA 建议开发对 蛋白质的非 Fc 区具有专属性的检测法。
3. 单克隆抗体
一些特殊的考虑因素涉及抗 mAb 抗体的检测。动物来源的 mAb,特别是啮齿动物来源的 mAb,预期 具有免疫原性,其中的免疫应针对整个 mAb 分子。
在治疗性 mAb 行业的早期,这是临床试验失败的一个关键原因(Kuus-Reichel, Grauer, et al.1994)。
减少 mAb 中非人序列存在的技术,例如嵌合和人源化,已经导致免疫原性显著降低,但不能消除免 疫原性 。在这些情况下,免疫应答主要针对 mAb 的可变区(Harding, Stickler, et al.2010; van Schouwenburg, Kruithof, et al.2014)。由于预期针对人 mAb 可变区的免疫应答,FDA 认为人 mAb 的使 用将不会进一步大幅度降低免疫原性。可以检测针对可变区的反应性的检测法被认为更适合于评价针 对基于 mAb 的治疗剂的抗体在患者中的潜在影响。然而,人抗体中 Fc 部分的工程改造(例如,非岩 藻糖基化水平的修饰)可能影响免疫原性。许多这些关注也涉及含有人 Fc 区的 Fc 融合蛋白。
4. 偶联蛋白
因为抗体-药物偶联物(ADC)是与小分子药物偶联的抗体,它们代表典型的半抗原载体分子。因此, 免疫原性检测法应当能够测量对 ADC 治疗性蛋白制品的所有组分的应答,包括抗体、连接子-药物和 可能由偶联产生的新表位。当需要对 ADC 进行标记用于免疫原性检测法时,应该仔细进行偶联,因 为已对 ADC 进行修饰。标记分子的疏水性潜在增加可能引起聚集,因此应该充分表征这些捕获试剂 的稳定性和溶解性。
5. 具有多个功能结构域的产品
一些蛋白质具有以不同方式起作用以介导临床有效性的多个结构域。对一个结构域的免疫应答可能抑 制特定功能,同时保留其他结构域完整。检查对具有多个功能结构域的治疗性蛋白制品的免疫应答可 能需要开发多种检测法以测量对分子的不同结构域的免疫应答。
V. 检测法开发
在下面的 A 至 D 节中提供了相应检测法类型开发的特定信息。这些章节补充与第 IV 节中提供的所有 检测法类型相关的信息。
A. 筛选检测法的开发
基于先前在 IV.A 节中讨论的多层方法,用于检测 ADA 的首选检测法应当是检测低亲和力和高亲和力 ADA 的高灵敏度筛选检测法。
大约 10 份单份样品可能用于在检测法开发的早期估计切点;然而,当目标人群的初治样品可用时, 这可能需要调整。低但定义的假阳性率对于初始筛选检测法是需要的,因为它使真阳性的检测最大化。 当确定免疫原性的真实发生率时,可以采用后续检测法来排除假阳性结果。
B. 确证性检测法的开发
因为筛选测定法旨在广泛检测在具有定义的假阳性率的血清样品中结合产品的抗体的存在,所以 FDA 建议申办者开发检测法以确认治疗性蛋白制品特异性抗体的结合。实施合适的确证性检测法对于防止 ADA 假阳性患者的数据混淆 ADA 对安全性和有效性的影响的分析是重要的。
1. 确证性检测法的格式选择
预期所选择的确证性检测法将至少与筛选检测法一样灵敏,但具有更高的专属性和至少一致的选择性 以鉴定任何假阳性样品。所选择的方法和仪器平台可能与用于筛选检测法的那些相似或不同。通常, 筛选检测法和确证性检测法使用相同的方法和仪器平台。在这种情况下,每种检测法的灵敏度将需要 以质量单位确定并且使用系统适用性对照确认,以确保检测法对结合抗体的存在是灵敏的。当使用结 合竞争检测法时,应当优化竞争产品的浓度以确认检测法范围之内和之上抗体的存在。
2. 确证性检测法的切点
如果选择竞争性抑制格式,则推荐用于确定切点的方法使用在竞争剂(通常是治疗性蛋白制品)存在 下来自抗体阴性初始患者样品的结合的数据。在这种情况下,用于建立切点的治疗性蛋白制品的量应 当与在检测法中用作竞争性抑制剂的治疗性蛋白制品的量相同。然而,这种方法可能不适合处理初始 人群中存在预先存在的抗体的样品。在这些情况下,申办者应该从切点评估中排除真阳性样品。在罕 见的情况下,当基线阴性样品不可用时,申办者可能评价滴度的变化或使用正交方法来确认筛选阳性 的样品。
C. 滴定检测法的开发
1. 滴度测定
滴度定义为样品给出筛选切点以上的值时的最大稀释度。滴度通常可以提供信息,并且可以与 ADA 的临床影响相关。
当患者具有预先存在的抗体时,滴定检测法可以提供特别信息。滴定检测法最经常使用与筛选检测法 相同的平台进行。通过连续稀释法检测血清。或者,可通过使用剂量反应曲线的线性部分将稀释度外 推至检测法切点来确定滴度。
2. 滴定检测法的切点
当患者具有预先存在的 ADA 时,可以通过治疗后滴度增加来鉴定治疗加强的 ADA 应答。需要用于 定义治疗后出现的或治疗加强的应答的切点。通常,该切点被确定为滴度,即当使用两倍稀释法来确 定滴度时,大于治疗前滴度的两倍稀释步骤。如果通过将稀释曲线外推至检测法切点确定滴度,则可 以使用检测法变异性的估计来确定治疗后出现的应答。
D. 中和检测法的开发
体外中和检测法提供了 ADA 抑制产品的生物活性的潜力的指示。这种 NAb 可以通过阻止产品到达其 靶标或通过干扰受体-配体相互作用而干扰治疗性蛋白制品的临床活性。选择用于评估 ADA 阳性样品 的中和潜力的检测方法应基于治疗性蛋白制品的作用机制。
1. 中和检测法的格式选择
已经使用两种格式的检测法来测量 NAb 活性:基于细胞的生物检测法和基于非细胞的竞争性配体结 合检测法。适当检测法格式的选择取决于各种因素。这些因素包括但不限于治疗性蛋白制品的作用机 制、其最接近地反映体内情况的能力以及检测法的选择性、灵敏度、精密度和耐用性。FDA 建议中和 检测法使用基于细胞的生物检测法格式,这取决于治疗性蛋白制品的作用机制,因为通常基于细胞的 生物检测法更密切地反映体内情况,并因此提供比配体结合检测法更相关的信息。因为基于细胞的生 物检测法通常基于产品的效价,所以历史上这些检测法的格式是极其可变的。效价生物检测法的选择 和设计通常基于细胞系对所讨论的产品应答的能力以及效价生物检测法与治疗性蛋白制品的作用机 制的相关性。
在这些生物检测法中测量的细胞应答是多种的,并且可以包括诸如细胞内底物的磷酸化、钙动员、增 殖和细胞死亡等结果。在一些情况下,申办者已经开发了表达相关受体或报道分子构建体的细胞系。 当治疗性蛋白制品直接刺激细胞应答时,可以测量 NAb 对生物检测法中生物活性降低的直接作用。 当治疗性蛋白制品间接影响细胞活性;例如通过阻断受体-配体相互作用,可以测量 NAb 对生物检测 法中生物活性恢复的间接作用。通常,生物检测法具有显著的变异性和其活性曲线的动态范围有限。 这些问题可能使得难以进行中和检测法的开发和验证。
存在可能使用配体结合检测法格式的情况。一种这样的情况是当不能开发具有足够灵敏度或选择性的 基于细胞的生物检测法时。另一种情况是当治疗性蛋白制品不具有基于细胞的作用机制时;例如靶向 血清蛋白的酶治疗性蛋白制品。配体结合检测法也可能适用于结合血清配体从而防止它们与其受体相 互作用的治疗性蛋白制品。然而,基于细胞的生物检测法可能仍然更适合于这种治疗性蛋白制品以证 明 ADA 抑制细胞活性。在这种情况下,申办者应与 FDA 讨论使用配体结合检测法。
2. 中和检测法的活性曲线
在检查中和检测法验证的其他要素之前,申办者应该仔细考虑剂量反应曲线(产品浓度与活性)。具 有小动态范围的检测法经证明可能不能用于测定中和活性。通常,中和检测法将采用单一浓度的治疗 性蛋白制品与单一稀释度的抗体。因此,申办者应选择活性读数对抑制灵敏的治疗性蛋白制品浓度。 如果检测法在接近剂量-反应曲线的平台(在下面的图 1 中标记为“否”)的浓度下进行,则可能不可能 鉴别具有低量 NAb 的样品。FDA 建议在曲线线性范围(图 1 中标记为“是”)的治疗性蛋白制品浓度 下进行中和检测法。该检测法还应该给出可重现的结果。
图 1. 代表性治疗性蛋白制品的活性曲线
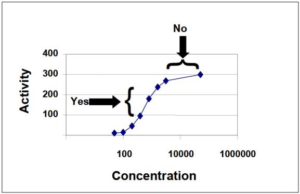
x 轴(浓度)表示治疗性蛋白制品的浓度,y 轴(活性)表示所得活性;例如,用治疗性蛋白制品刺 激后细胞系的细胞因子分泌浓度。该曲线显示对治疗性蛋白制品的陡峭反应,其在约300 时达到平台。 “否”箭头表示不适合在单剂量中和检测法中使用的治疗性蛋白制品的浓度,因为它代表由治疗性蛋白 制品诱导的活性对于 NAb 的抑制相对不灵敏的浓度范围。“是”箭头表示曲线的线性部分上的由治疗 性蛋白制品诱导的活性对于 NAb 的中和灵敏的浓度范围。
3. 中和检测法的基质干扰的考虑因素
基质可以引起对中和检测法的干扰,特别是因为血清或血浆组分可以在生物检测法中增强或抑制治疗 性蛋白制品的活性。例如,来自患有特定疾病的患者的血清可能含有升高水平的一种或多种细胞因子, 这些因子可能用于激活生物检测法中的细胞,并通过增加对原始刺激因子或治疗性蛋白制品的应答而 掩盖 NAb 的存在。因此,申办者应该理解这些检测法中的基质效应。富集血清或血浆样品中 ADA 等 方法可能适用于这些类型的情况。然而,这种方法可能导致 NAb 的损失,因此需要由申办者仔细检 查和验证。
在中和检测法中使用的治疗性蛋白制品的浓度对检测法灵敏度具有关键影响。FDA 认识到尽管使用低 浓度的治疗性蛋白制品可导致对抗体的抑制更灵敏的中和检测法,但非常低浓度的治疗性蛋白制品可 能导致检测法的精密度不佳。关于基质干扰的一般信息,请参见第 IV.D.1 节。
4. 中和检测法的切点
确定检测法切点曾经对中和检测法提出了巨大的挑战。与所有检测法一样,切点应基于使用来自初治 受试者的样品建立的检测法变异性来确定。如果对在筛选和验证检测法中检测为阳性的样品进行中和 检测,则 1%假阳性率是可接受的。如果使用中和检测法进行筛选,应使用 5%的假阳性率(参见第 VI.B.2 节)。如果样品变异性程度使得难以评估 NAb 活性,可以考虑其他方法,但应在实施前与 FDA 讨论。或者,探索导致较小变异性并提供更准确的切点指定的其它检测法格式可能是必要的。关于检 测法切点的一般信息,同样请参见第 IV.B 节。
5. 中和检测法的其他考虑因素
因为中和检测法最常见地仅在经确认具有抗原特异性 ADA 的样品上进行,所以通常不需要确证性方 法。然而,由于生物检测法的复杂性,检测法专属性的确认可能可用于确定患者是否具有真正的 NAb 应答。申办者应考虑以下方法:
- 不相关的抑制性分子可引起中和活性,有时可能不清楚所观察到的中和活性是否由中和抗体或其他抑制性分子引起。来自基线暴露前样品的检测结果可能提供信息。当担心存在非特异性抑制时,应进行抗体耗竭检测法以评价中和活性是否真正由 ADA 引起,而不是由其他抑制性分子引起。
- 细胞系可以对除所研究的治疗性蛋白制品之外的多种刺激产生应答。在这种情况下,可以在存在 治疗性蛋白制品(应当被特异性NAb 应答阻断)或替代刺激(应当不会被特异性NAb 应答阻断)的 情况下检查 NAb 的存在。
- 血清可能包含可能在中和检测法中产生假结果的组分,例如可溶性受体或内源配对物。
在这种情况下,在不存在治疗性蛋白制品的情况下将检测血清或血浆样品直接加入生物检测法可能可 用于理解检测法结果。
VI. 检测法验证
检测法验证是通过使用特定的实验室研究证明所使用的 ADA 检测法的性能特征适合其预期用途的过 程。23 验证水平取决于产品开发的阶段和与治疗性蛋白制品相关的患者的免疫原性的结果的风险。涉 及评估测定灵敏度、专属性和精密度要求而较少强调耐用性、重现性和稳定性的部分验证可能足以用 于临床开发的早期阶段,例如 I 期和 II 期研究。然而,作为一个科学问题,如第 VI.A 节所述,应该 对关键性研究和上市后研究使用经完全验证的检测法。
第 VI.B 至 VI.E 节中提供了相应检测法类型验证的特定信息。这些章节补充了与第 IV 节和第 VI.A 节 提供的所有检测法类型相关的信息。
A. 检测法验证的一般考虑因素
应使用经完全验证的检测法对来自关键性研究的样品进行检测。在许可申请时,申办者应提供支持该 检测法的全面验证的数据。验证包括证明用于给定样品中 ADA 的定量测量的特定检测法对于预期用 途是可靠和可重现的所有程序。验证的基本参数包括(1)切点,(2)灵敏度,(3)专属性和选择性, (4)精密度,(5)相关时的重现性,(6)某些检测法特征的稳定性以及试剂和对照样品的稳定性。 由检测法产生的临床数据的可接受性直接对应于用于验证检测法的标准。
检测法切点是检测法验证的一个基本方面。如果来自适当患者人群的初治样品不能用于研究前验证活 动,则可以使用替代样品。通常这些是来自商业来源的样品。当使用替代样品来确定验证活动中的切 点时,一旦来自合适人群(例如初治患者)的样品可用,就应当再次确定切点。使用适当样品验证的 切点应用于确定样品是否呈 ADA 阳性。
为了验证基本检测法参数,FDA 建议至少在至少 3 个不同天评价检测法间精密度,2 名分析员,每名 制备相同样品的至少另外 6 份独立制备物,使用相同的仪器平台和型号。
23 参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗体的免疫检测法的设计和验证。同样参见生物分析方法验证行业 指南、USP 通则 1225 药典方法验证以及 ICH 行业指南 Q2(R1)分析方法验证:正文和方法学。
应该用同一分析员独立制备的每板相同样品的至少 6 份独立制备物来评价检测法内精密度。在检测法 内精密度或检测法间精密度具有大于 20%的变异系数(%CV)的情况下,申办者应考虑改进检测法 参数以便尽可能优化检测法精密度的需要或提供解释为什么更高的%CV 应该是可接受的的理由。或 者,当在板上不可能运行相同样品的 6 份独立制备物时,在具有低通量的检测法(例如,滴度检测法) 中,应当用每板相同样品的至少 3 份独立制备物和相同样品的至少总共 9 份独立制备物来评价检测法 内精密度。样品应包括其检测产生检测法动态范围的低、中和高水平值的阴性对照和阳性样品。申办 者应在相关时评价仪器间和操作员间精密度。在相同的操作条件下,检测法在不同操作员之间应具有 相当的精密度。
当对先前已验证的方法进行变更时,申办者应该对需要多少额外的验证进行判断。在典型的产品开发 计划过程中,可能对所定义的 ADA 检测法进行修饰。偶尔,样品可能需要用优化的经验证检测法重 新检测;因此,应当规定在允许重新检测的条件下保存足够的样品体积,直到检测法经监管机构完全 验证和评价。24
应验证关键方法参数,例如孵育时间和温度,以证明检测法在这些参数的预定范围内按照预期进行。 通常,在验证活动中检测允许范围的低、中和高值。
根据用于检测法的方法(或技术)和仪器平台,可能需要验证其他参数。例如,应当验证表面等离子 体共振检测法在再生时的表面稳定性,并且应当针对芯片的基线性能设置标准。应当确定标记25试剂 的效率和稳定性。申办者应在开发阶段检查耐用性,如果检测法中特定步骤的微小变化影响结果,则 应采取特定预防措施来控制其变异性。
24 关于不同类型和水平的验证,参见生物分析方法验证行业指南。另外参见 USP 通则 1106 免疫原性检测法-用于检测抗药 物抗体的免疫检测法的设计和验证。
25 如果试剂与将有助于其捕获或可视化的部分偶联或融合,则认为该试剂被标记;例如,与生物素、链霉亲和素或荧光染 料偶联。未标记试剂是未标记的试剂(例如,药物)。
B. 筛选检测法的验证
1. 筛选检测法的灵敏度
先前讨论的检测法验证的所有一般考虑因素适用于筛选检测法的验证。如前所述,灵敏度在初始筛选 检测法中特别重要,因为这些结果决定了样品的进一步分析。
2. 筛选检测法的切点
应由至少 2 名分析员在至少 3 个不同天检测至少 50 份样品来统计学确定切点,使用合适的统计方法。 FDA 建议筛选检测法的切点由阴性对照人群的第 95 百分位数的 90%单侧置信区间下限确定(Shen, Dong, et al.2015)。这将保证至少 5%的假阳性率以及 90%的置信水平。这种方法提高了鉴别所有可能 产生抗体的患者的检测法的概率。用于确定切点的统计方法应基于数据的统计分布。例如,正态分布 的第 95 百分位数由平均值加上 1.645 标准偏差估计。其他方法可能用于估计第 95 百分位数,包括使 用中值和中值绝对偏差值代替平均值和标准偏差。
阴性对照样品的平均响应可能是恒定的或可能在检测法、平板或分析物之间变化。当平均值恒定时, 可能在可以应用于研究中的检测法的检测法验证期间确定切点。这通常被称为固定切点。当平均值在 检测法、平板或分析物之间变化但是围绕平均值的方差是恒定的时,可以统计学确定归一化因子并应 用于研究中。这也被称为浮动切点。当平均值和方差都变化时,必须针对每个检测法、平板或分析物 确定切点。这被称为动态切点。动态切点的一个缺点是需要在检测法中具有更多的阴性对照的重复。 动态切点不应用于补偿缺陷检测法优化。
C. 确证性检测法的验证
确证性检测法应该以类似于筛选检测法和中和检测法的方式完全验证,因为这些检测法提出一些特定 问题。作为一个科学问题,确证性检测法的研究将取决于所选择的检测法格式和仪器。如果这些检测 法基于患者样品中抗体对抗原结合26 的竞争,并且测量指标是响应丧失,则关键是确定将用于将阳性 归因于样品的抑制或耗竭的程度。在过去,使用结合减少的固定百分比,但这些数字通常是任意的, 并且不可能与所有检测法相关。
26 抗原结合的竞争是指竞争检测法,其中抗原特异性抗体结合标记的或平板结合的抗原的能力被未标记或可溶性抗原抑制。
FDA 建议基于在加入竞争抗原时已知缺乏抗体的样品中观察到的结合变化的评估来确定切点。FDA 还建议使用低浓度的阳性对照抗体来确认确证性检测法的灵敏度。
对于确证性检测法切点的估计,推荐第 99 百分位数的 80%单侧置信区间下限。因为这种检测法的目 的是消除由于非特异性结合而产生的假阳性样品,所以使用 1%假阳性率计算确证性切点是足够的。 不推荐使用更严格的假阳性率(如 0.1%),因为它会导致假阴性结果的风险增加。关于检测法切点的 一般信息,请参见第 IV.B 节。
如果确证性检测法格式是竞争性检测法,其中将竞争剂(通常是未标记的治疗性蛋白制品)27添加到 反应混合物中以抑制 ADA 与用于切点检测法的捕获试剂结合,当确定确证性切点时,应当向样品中 加入相同浓度的未标记的治疗性蛋白制品。
D. 滴定检测法的验证
第 VI.A 节中描述的检测法验证的原则通常适用于滴定检测法的验证。滴定检测法的切点可能与筛选 检测法的切点相同或不同。当滴定检测法不用于筛选并且切点不同于筛选检测法的切点时,可能需要 验证单独的滴定方法切点;例如当来自检测法稀释剂或基质的信号由于血清的阻断效应而导致比筛选 检测法切点更高的结果时,或者如果高于 MRD 的稀释度样品不产生始终如一的阴性结果,即当筛选 切点落在阳性对照稀释曲线的下限平台上。28
E. 中和检测法的验证
应由至少 2 名分析员在至少3 个不同天检测至少30 份样品来确定切点,使用合适的统计方法。
FDA 认为不是所有的 ADA 都是中和的,并且可能难以鉴别具有中和能力的阳性对照抗体。此外,如 果使用亲和纯化的多克隆阳性对照抗体制剂,则可能仅有一部分抗体是中和的,这可使检测法看起来 不太灵敏。因此,重要的是验证检测法灵敏度。
申办者应验证基于细胞的中和生物检测法的检测法专属性。
27 参见脚注 25。
28 参见 USP 通则 1106 免疫原性检测法-用于检测抗药物抗体的免疫检测法的设计和验证。
如上所述,对于可能响应于除特定治疗性蛋白制品以外的刺激的细胞,证明 NAb 仅抑制对治疗性蛋 白制品的应答而不抑制对其他刺激的应答的能力是检测法专属性的良好指示。在这些研究中,FDA 建 议以产生类似于治疗性蛋白制品的结果的浓度使用其他刺激。申办者还应确认患者血清中不存在替代 刺激(参见第 IV.C 节和 IV.D 节)。
与配体结合检测法相比,基于细胞的中和生物检测法通常具有降低的精密度,因为生物应答可以比仔 细控制的结合研究本身更可变。因此,与筛选检测法相比,申办者应当进行更多的重复以评估精密度 和评估患者应答(参见第 IV.E 节)。
当使用在允许传代次数、细胞密度和细胞活力的低、中和高范围的细胞时(参见第 IV.G 节),应当验 证的其他参数是检测法性能。
VII. 检测法检测的实施
A. 获得患者样品
FDA 建议申办者从所有患者获得暴露前样品。因为在基质中有存在预先存在的抗体或混杂组分的潜力, 所以了解治疗之前的反应性程度是必要的。申办者应获得随后的样品,采样时间取决于给药频率。最 佳地,在第一次暴露后 7 至 14 天采集的样品可以帮助阐明早期 IgM 应答。在第一次暴露后 4 至 6 周 采集的样品通常对于测定 IgG 应答是最佳的。对于接受治疗性蛋白制品单次给药的个体,上述时间框 架可能是足够的。然而,对于在试验期间多次接受治疗性蛋白制品的患者,申办者应在整个试验期间 以适当的时间间隔获得样品,并且在最后一次暴露后约 30 天获得样品。
在血清中存在的治疗性蛋白制品产生最小干扰时获取样品是必需的。申办者应考虑治疗性蛋白制品的 半衰期,以帮助确定适当的采样时间。这对于 mAb 产品特别重要,因为这些产品可以具有几周或更 长的半衰期;并且取决于给药方案,治疗性 mAb 本身可以在血清中保持存在数月。在需要检测 IgE 的情况下,应与 FDA 讨论样品采集时间。
干扰检测法的治疗性蛋白制品水平(通过免疫竞争测定)还可以帮助定义有意义的采样时间点。如果 在试验的治疗阶段期间不能获得不含治疗性蛋白制品的样品,则申办者应在适当的清除期后(例如 5 个半衰期)采集其他的样品。如果所讨论的治疗性蛋白制品本身是免疫抑制剂,获得样品以检测有意 义的抗体结果也可能是复杂的。
在这种情况下,申办者应该从已经经历洗脱期的患者获得样品,因为治疗期已经结束或因为患者已经 退出研究。
用于测定治疗性蛋白制品的血清浓度的样品应当与免疫原性样品同时获得。检测这类样品可以提供关 于在样品中的治疗性蛋白制品是否干扰 ADA 检测和 ADA 是否可能改变治疗性蛋白制品的药代动力 学的信息。
B. 并行阳性和阴性质量对照
如果申办者完成正确的验证工作并进行切点确定,则应该直接确定患者的免疫原性状态。然而,阳性 对照或 QC 样品是关键的,并且应当与患者样品同时运行。我们建议这些样品跨越在检测法中具有已 知的负、低和高反应性的 QC 样品的阳性水平。更重要的是,QC 样品应在检查患者样品的基质中稀 释;例如相同百分比的血清或血浆(指定使用的盐抗凝剂)。以这种方式,申办者确保检测法正在执 行其最佳准确度,并且正确地评价患者样品。对于低阳性 QC 样品,我们建议选择经统计分析将导致 1%时间的检测法运行拒收的浓度。当对实际患者样品进行时,这种方法将确保检测法的适当灵敏度。 应设定高阳性 QC 样品的浓度以监测前带效应。29
FDA 还建议从具有由第二检测试剂检测的抗体的人或动物获得这些 QC 样品,以确保可能观察到的阴 性结果真正由缺乏抗原反应性引起,而不是由第二试剂的失效引起。这个问题不是抗原桥接检测法的 问题,因为使用标记抗原进行检测。
C. 确认目标人群的切点
来自不同人群的样品在 ADA 检测法中可具有不同的背景活性。因此,有必要确认在检测法验证期间 确定的切点适用于所研究的人群体。类似地,如果未以代表如何在研究中获得和处理样品的方式获得 和处理用于在检测法验证期间切点确定的样品,则也应该用适当的样品来确定切点。应使用来自目标 人群的足够数量的样品,并应提供所使用数量的理由。如果没有足够数量的样品,应与监管机构商定 使用的样品数量。
29 前带效应(也称为钩状效应)是由于存在高浓度的特定分析物或抗体而可能发生的信号的减少,并且可能导致假阴性结果。
VIII. 文件
免疫原性检测模式的理由和信息应在关于人类研究的生物分析和分析方法报告的电子通用技术文件 (eCTD)模块 5.3.1.4 中提供。30 应将使用的相应检测法的标准操作规程与验证研究的结果和未验证 的参数(例如 MRD、在 Nab 检测法中使用的治疗性蛋白制品的刺激浓度以及对检测法性能至关重要 的一些耐用性参数)的相关检测法开发信息提供给 FDA(参见生物分析方法验证行业指南草案中的第 VII 节文件)。31
30 关于 eCTD 申报资料的更多信息,请访问 FDA 网站,网址:
http://www.fda.gov/Drugs/DevelopmentApprovalProcess/FormsSubmissionRequirements/ElectronicSubmissions/uc m153574.htm. 关于将要提交给监管机构的申报资料的 CTD 结构良好有效性部分编制的商定通用格式的更多信息,请参阅ICH 行业指南 M4E:CTD-有效性。关于申办者和申请人如何根据 FD&C 法案第 745A(a)节组织他们电子提交给监管机构的所有申报资 料类型的内容的详细信息,请参阅行业指南(及其通过引用并入的技术质量标准文件)采用eCTD 规范编制电子格式注册申 报资料—某些人用药品申请和相关申报资料行业指南。
31 最终定稿后,本指南代表 FDA 目前关于该问题的想法。为了确保您拥有最新版本的指南,检查 FDA 指南网页 http://www.fda.gov/RegulatoryInformation/Guidances/default.htm。
参考文献
Aalberse, R. C. and J. Schuurman (2002). “IgG4 breaking the rules.” Immunology 105(1): 9-19. Boes, M. (2000). “Role of natural and immune IgM antibodies in immune responses.” Mol
Immunol 37(18): 1141-1149.
Calabresi, P. A., G. Giovannoni, et al.(2007). “The incidence and significance of anti- natalizumab antibodies: results from AFFIRM and SENTINEL.” Neurology 69(14): 1391-1403.
Caruso, A. andA. Turano (1997). “Natural antibodies to interferon-gamma.” Biotherapy 10(1): 29-37.
Cohen, B. A. and V. M. Rivera (2010). “PRISMS: the story of a pivotal clinical trial series in multiple sclerosis.” Curr Med Res Opin 26(4): 827-838.
Coutinho, A., M. D. Kazatchkine, et al.(1995). “Natural autoantibodies.” Curr Opin Immunol 7(6): 812-818.
Disis, M. L., V. Goodell, et al.(2004). “Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients.” J Clin Immunol 24(5): 571-578.
Goodin, D. S., E. M. Frohman, et al.(2007). “Neutralizing antibodies to interferon beta: assessment of their clinical and radiographic impact: an evidence report: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology.” Neurology 68(13): 977-984.
Gupta, S., V. Devanarayan, et al.(2011). “Recommendations for the validation of cell-based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics.” J Pharm Biomed Anal 55(5): 878-888.
Gupta, S., S. R. Indelicato, et al.(2007). “Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics.” J Immunol Methods 321(1-2): 1-18.
Harding, F. A., M. M. Stickler, et al.(2010). “The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions.” MAbs 2(3): 256-265.
Hintermann, E., M. Holdener, et al.(2011). “Epitope spreading of the anti-CYP2D6 antibody response in patients with autoimmune hepatitis and in the CYP2D6 mouse model.” J Autoimmun 37(3): 242-253.
Kuus-Reichel, K., L. S. Grauer, et al.(1994). “Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? ” Clin Diagn Lab Immunol 1(4): 365- 372.
Matsumoto, T., M. Shima, et al.(2001). “Immunological characterization of factor VIII autoantibodies in patients with acquired hemophilia A in the presence or absence of underlying disease.” Thromb Res 104(6): 381-388.
Miller, L. L., E. L. Korn, et al.(1999). “Abrogation of the hematological and biological activities of the interleukin-3/granulocyte-macrophage colony-stimulating factor fusion protein PIXY321 by neutralizing anti-PIXY321 antibodies in cancer patients receiving high-dose carboplatin.” Blood 93(10): 3250-3258.
Mire-Sluis, A. R., Y. C. Barrett, et al.(2004). “Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products.” J Immunol Methods 289(1-2): 1-16.
Plotkin, S. A. (2010). “Correlates of protection induced by vaccination.” Clin Vaccine Immunol 17(7): 1055-1065.
Prummer, O. (1997). “Treatment-induced antibodies to interleukin-2.” Biotherapy 10(1): 15-24.
Ramsland, P. A., B. F. Movafagh, et al.(1999). “Interference of rheumatoid factor activity by aspartame, a dipeptide methyl ester.” J Mol Recognit 12(5): 249-257.
Ross, C., M. B. Hansen, et al.(1990). “Autoantibodies to crude human leucocyte interferon (IFN), native human IFN, recombinant human IFN-alpha 2b and human IFN-gamma in healthy blood donors.” Clin Exp Immunol 82(1): 57-62.
Shankar, G., V. Devanarayan, et al.(2008). “Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products.” J Pharm Biomed Anal 48(5): 1267-1281.
Shen, M., X. Dong, et al.(2015). “Statistical evaluation of several methods for cut-point determination of immunogenicity screening assay.” J Biopharm Stat 25(2): 269-279.
Thrasyvoulides, A., E. Liakata, et al.(2007). “Spreading of antibody reactivity to non-thyroid antigens during experimental immunization with human thyroglobulin.” Clin Exp Immunol 147(1): 120-127.
Turano, A., A. Balsari, et al.(1992). “Natural human antibodies to gamma interferon interfere with the immunomodulating activity of the lymphokine.” Proc Natl Acad Sci U S A 89(10): 4447-4451.
van der Meide, P. H. and H. Schellekens (1997). “Anti-cytokine autoantibodies: epiphenomenon or critical modulators of cytokine action.” Biotherapy 10(1): 39-48.
van der Woude, D., S. Rantapaa-Dahlqvist, et al.(2010). “Epitope spreading of the anti- citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis.” Ann Rheum Dis 69(8): 1554-1561.
van Schouwenburg, P. A., S. Kruithof, et al.(2014). “Functional analysis of the anti-adalimumab response using patient-derived monoclonal antibodies.” J Biol Chem 289(50): 34482-34488.
Zhou, L., S. A. Hoofring, et al.(2013). “Stratification of antibody-positive subjects by antibody level reveals an impact of immunogenicity on pharmacokinetics.” AAPS J 15(1): 30-40.
Hits: 150
- 心脏病成人的疫苗接种指南
- 用于检测治疗性蛋白制品的免疫原性的检测法开发和验证行业指南
- 肺部疾病成人的疫苗接种指南
- 快速响应:截至 2024 年 12 月在非大流行背景下人类接种禽流感疫苗的初步指南
- 糖尿病成人的疫苗接种指南
- 慢性肝病或慢性肝脏感染成人的疫苗接种指南
- 人类免疫缺陷病毒(HIV)感染者成人疫苗接种指南
- 65 岁及以上成人流感疫苗接种补充指南
- 2013 年美国感染病学会(IDSA)免疫功能低下人群疫苗接种临床实践指南
- 针对公共卫生紧急情况的猴痘检测政策
- 卫生保健机构手部卫生指南
- 环境感染控制指南 保健设施
- 2025 年 4 月 30 日的 NACI 声明摘要:关于 2025-2026 年季节性流感疫苗的声明
- 更新了老年人呼吸道合胞病毒 (RSV) 疫苗指南,包括扩大 RSVPreF3 在 50-59 岁人群中的使用以及使用新的 mRNA-1345 疫苗
- 孕期梅毒感染筛查:美国预防服务工作组重申建议声明
- 2025 年 5 月 14 日的 NACI 声明摘要:关于免疫功能低下成人带状疱疹疫苗接种的最新建议
- 2022 美国动物卫生协会犬类疫苗接种指南(2024年更新)
- 2020 年 AAHA/AAFP 猫科动物疫苗接种指南
- 2025 年 5 月 15 日的 NACI 声明摘要:关于使用 Imvamune 预防 mpox 的更新指南的快速响应
- 2025 年 2 月 13 日 NACI 声明摘要:关于麻疹暴露后预防的更新建议
- 从 2025 年 7 月 1 日起完成常规免疫接种计划(英国)
- 2025 年 1 月 10 日的 NACI 声明摘要:2025 年至 2026 年夏季 COVID-19 疫苗使用指南
- 麻疹暴露后预防更新建议的增编
- 使用 Imvamune 预防猴痘的更新指南
- 免疫功能低下成人带状疱疹疫苗接种的最新建议
- 推荐使用帕利珠单抗减少婴儿呼吸道合胞病毒感染的并发症
- 登革热防控方案(2025年版)
- 关于防止节肢动物叮咬的个人防护措施的声明 – 更新
- CATMAT 关于在 65 岁或以上人群中使用基孔肯雅热减毒活疫苗 (IXCHIQ) 的指南
- 关于在授权旅行者使用 QDENGA(登革热疫苗)的司法管辖区使用的建议
- 基于暴露的干预措施,用于管理整个生命周期中高度针头恐惧的个体:临床实践指南,呼吁进一步研究
- mRNA COVID-19 疫苗接种后患有心肌炎和心包炎的青少年的临床指南
- 归国国际旅客发热初步评估指南
- NACI 快速响应:在持续的猴痘疫情背景下更新了关于 Imvamune 的临时指南
- 关于管理耐多药沙门氏菌感染的临时指南
- 家长免疫接种指南
- 英国旅行者接种基孔肯雅热疫苗:JCVI 建议
- 针对 80 岁及以上成年人的呼吸道合胞病毒 (RSV) 免疫计划:JCVI 建议,2025 年 7 月 16 日
- JCVI 关于 2026年秋季和 2027 年春季 COVID-19 疫苗接种的声明
- CATMAT 关于播散性类圆线虫病的声明
- 成人免疫接种指南
- 针对呼吸道合胞病毒感染住院高风险婴幼儿的帕利珠单抗预防更新指南
- 针对呼吸道合胞病毒感染住院高风险婴幼儿的帕利珠单抗预防
- JCVI 关于 2026 年至 2027 年流感疫苗的声明
- 有关打算探亲访友的国际旅客的声明摘要
- 青少年疫苗接种指南
- 免疫接种状态不确定或不完全的个人疫苗接种:2025 年 7 月 1 日起
- 从 2025 年 7 月 1 日起进行常规儿童免疫接种(2024 年 7 月 1 日出生的婴儿)
- 加拿大关于在疫苗短缺期间使用分次剂量黄热病疫苗的临时建议
- 寨卡病毒预防和治疗建议
- 带状疱疹免疫接种计划:医护人员资讯
- 带状疱疹疫苗接种指南
- 儿童和青少年接种COVID-19疫苗
- 印度旅行疫苗接种:2025 年循证健康指南
- 旅行者疟疾预防和治疗指南
- 登革热:给明智旅行者与移居者的详尽旅行指南
- 旅行者伤寒:预防和保护完整指南
- 美国儿科学会 2025 年 8 月 19 日发布的推荐18 岁及以下儿童与青少年免疫接种程序
- 隐藏的敌人:旅行者立克次体感染完整指南
- 心脏病发作症状:旅行者的基本医疗指南
- 真菌性皮肤感染:类型、治疗和预防完整指南
- 热带医学和旅行咨询委员会 (CATMAT) 关于国际旅行者和伤寒的声明
- 2025 年简明临床指南:ACC 关于成人免疫接种作为心血管护理一部分的专家共识声明:美国心脏病学会解决方案集监督委员会的报告
- 产科-妇科护理中的 COVID-19 疫苗接种注意事项
- 妊娠期流感:预防和治疗
- 母体呼吸道合胞病毒疫苗接种
- 疫苗预防和控制季节性流感:免疫实践咨询委员会的建议——美国,2025-26 年流感季节
- 用Clesrovimab预防婴儿严重呼吸道合胞病毒相关下呼吸道感染:免疫实践咨询委员会的建议 — 美国,2025年
- 健康怀孕指南
- 关于预防婴幼儿呼吸道合胞病毒感染的建议:政策声明
- 过敏反应定义、概述和临床支持工具:2024 年共识报告 – GA2LEN项目
- 人狂犬病免疫球蛋白(human rabies immunoglobulin, HRIG)使用临时建议
- 如何与我们的心脏病患者讨论成人疫苗接种
- 2025 年秋季疫苗指南
- 疫苗佐剂非临床研究技术指导原则
- 完整的常规免疫接种计划 英国 2025年9月1日期
- 2025 年至 2026 年国家流感免疫接种计划信函
- 恰加斯病:基本旅行指南
- 创新型疫苗临床试验申请申报资料要求
- 远离结核病的旅行者指南
- 更顺畅的免疫接种体验指南
- 您的免疫预约指南:充满信心地完成整个过程
- 联合疫苗临床前和临床研究技术指导原则
- 疫苗接种预约指南:预期事项与准备方法
- 旅行时如何保护儿童免受水痘侵害:专家指南
- 家长互联网免疫接种信息指南
- 您的无缝免疫接种与健康服务指南
- 2025年病毒爆发与旅行警告:专家友好指南
- 结合疫苗质量控制和临床研究技术指导原则
- 预防用含铝佐剂疫苗技术指导原则
- 生物制品稳定性研究技术指导原则(试行)
- 常见传染病的暴露后预防
- 无缝接种预约指南
- 开发用于预防全球传染病的疫苗的一般原则
- 关于预防性疾病的联合疫苗评估:生产、测试和临床研究
- 关于组合疫苗及免疫学兽用药品(IVMPs)联合使用的指导原则
- 世界卫生组织关于疫苗非临床评价的指南
- 确保基于DT的组合疫苗的质量、安全和有效性的建议
- 沙特 疫苗生产和质量控制指南
- 疫苗接种预约指南:流程与准备事项
- 儿童与青少年疫苗接种时间指南
- 聚焦预防 | 美国心脏病学会简化心血管疾病免疫接种建议
- 精通疫苗接种诊所:提高效率与优化患者护理的内部指南
- 临床医生呼吸道疫苗共同临床决策指南
- 你的疫苗接种顺利体验指南
- 美国传染病学会关于A组链球菌性咽炎的临床实践指南更新
- 更顺畅疫苗接种体验终极指南
- 婴幼儿和青少年COVID-19疫苗接种建议:政策声明
- IDSA 2025年关于在免疫功能低下患者中使用疫苗预防季节性新冠病毒、流感和呼吸道合胞病毒感染的指南
- 无脾成人的疫苗接种指南
- 花生过敏预防的积极趋势:预防指南的实际影响
- 2025–2026年儿童流感预防与控制建议:技术报告
- 您的疫苗诊所指南:像专业人士一样导航系统
- 带状疱疹疫苗临床试验技术指导原则
- PIDS/IDSA 2023年儿童急性细菌性关节炎诊断和管理指南
- 预防节肢动物叮咬的个人防护措施声明
- 异维甲酸与疫苗诊所:保持防护的内部指南
- IDSA 2024年抗微生物药物耐药革兰氏阴性菌感染治疗指南
- 重组糖蛋白激素类产品药学研究与评价技术指导原则
- 流感病毒疫苗临床试验技术指导原则(征求意见稿)
- 重组表达单克隆抗体类生物制品制造及检定规程撰写指南(征求意见稿)
- 归国国际旅行者发热初始评估指南
- 中国流感疫苗预防接种技术指南(2025—2026)
- 成人癌症相关免疫抑制患者抗菌药物预防临床实践指南:ASCO/IDSA更新版
- 免疫功能低下旅行者
- 全国镰状细胞中心联盟关于镰状细胞病健康维护的共识建议共识声明
- 多联疫苗临床试验技术指导原则
- 曲霉病诊断和管理临床实践指南:IDSA 2016年更新版
- 养老院感染预防与控制多学会指南
- 关于跨国收养的声明
- 美国传染病学会 (IDSA) 发布的《念珠菌病管理临床实践指南:2016 年更新版》
- 流感疫苗完全指南
- IWGDF/IDSA糖尿病相关足部感染诊疗指南
- 热带病与旅行咨询委员会:老年旅行者声明
- 疫苗临床试验不良事件分级标准指导原则(修订版)
- ATS/CDC/IDSA临床实践指南:成人和儿童结核病的诊断
- 用于人乳头瘤病毒检测的自行收集阴道标本及筛查终止指南:美国癌症协会宫颈癌筛查指南更新
- 热带医学与旅行咨询委员会关于儿童旅行者的声明
- 成人导管相关性尿路感染的诊断、预防和治疗:美国传染病学会2009年国际临床实践指南
- HIVMA/IDSA 2014年更新版:HIV感染患者慢性肾病管理临床实践指南
- 对免疫状况不明或未完成的人员进行疫苗接种:自2026年1月1日起
- SHEA/IDSA 发布的《成人艰难梭菌感染管理临床实践指南:2021 年更新》
- 加拿大传染病报告:孕期旅行声明
- 成人和儿童艰难梭菌感染临床实践指南:美国医疗保健流行病学学会/美国感染病学会2017年更新版
- 关于旅行者与狂犬病疫苗的声明
- IDSA 2016 年球孢子菌病治疗临床实践指南
- 免疫接种者袖珍指南:孕期与哺乳期
- 成人社区获得性肺炎的诊断和治疗:美国胸科学会和美国传染病学会官方临床实践指南
- CATMAT 关于播散性粪类圆线虫病的声明:预防、评估和管理指南
- 3个月以上婴幼儿社区获得性肺炎的管理:儿科传染病学会和美国传染病学会临床实践指南
- 复杂性尿路感染(cUTI):治疗和管理临床指南
- 自行采集阴道标本用于人乳头瘤病毒检测及筛查退出指南:美国癌症协会宫颈癌筛查指南更新
- 旅行者结核病风险评估与预防
- IDSA 新型冠状病毒肺炎患者治疗和管理指南
- 热带病与旅行咨询委员会(CATMAT) 致旅行者及关于黄热病的声明
- 预防用疫苗佐剂药学研究技术指导原则(试行 )
- 隐球菌病治疗临床实践指南: 美国感染病学会2010年更新
- 儿童常规免疫接种计划
- ASM/ECMM/ISHAM 隐球菌病诊断和治疗全球指南
- 加拿大各省及地区婴幼儿常规(及补种)疫苗接种时间表(截至2025年12月)
- 美国儿科学会推荐的青少年及年轻成人疫苗
- IDSA 脑炎管理指南
- 加拿大各省和地区针对健康且先前已接种疫苗的成年人的常规疫苗接种计划
- 美国2026年儿童和青少年免疫接种计划建议
- 美国心脏协会成人感染性心内膜炎指南:诊断、抗菌治疗和并发症处理
- IDSA 2008 年老年长期护理机构居民发热和感染评估临床实践指南更新
- 癌症成人患者的疫苗接种:ASCO指南
- 癌症患者治疗前的乙型肝炎病毒筛查与管理:美国临床肿瘤学会临时临床意见更新
- CEPI 生物安全政策
- IDSA 关于 A 组链球菌(GAS)咽炎的临床实践指南更新
- 2026 年美国 18 岁及以下儿童和青少年推荐免疫接种程序
- 2025-2030 年美国人膳食指南
- AASLD/IDSA 2023 年丙型肝炎病毒感染检测、管理和治疗临床实践指南更新
- 关于接受度普利尤单抗治疗的患者使用疫苗的系统评价和专家德尔菲共识建议:美国过敏、哮喘和免疫学会立场文件
- 常规推荐的孕产妇疫苗接种汇总
- SHEA/APIC/IDSA/PIDS 多学会联合立场文件:提高标准——有效医疗机构感染预防和控制计划所需的资源和结构
- 美国医疗机构中感染乙型肝炎、丙型肝炎或人类免疫缺陷病毒的医护人员管理办法
- 医护人员疫苗接种咨询四步法指南
- IDSA 2017 年医疗相关性脑室炎和脑膜炎临床实践指南
- AASLD ISDA 慢性乙型肝炎治疗实践指南
- 美国传染病学会 2025 年组织胞浆菌病临床实践指南更新:成人、儿童和孕妇无症状组织 胞浆菌肺结节(组织胞浆菌瘤)及轻度或中度急性肺组织胞浆菌病的治疗
- 呼吸中心:您的 GoodRx 指南
- 2017 年 HIVMA/IDSA 艾滋病毒感染者慢性疼痛管理临床实践指南
- 人乳头瘤病毒疫苗临床试验技术指导原则 (修订版)
- 流感病毒疫苗临床试验技术指导原则(试行)
- 美国传染病学会和美国胸科学会2016年临床实践指南:成人医院获得性肺炎和呼吸机相关性肺炎的管理
- 美国内科医师学会免费提供的 2025-2026 年成人新冠疫苗接种快速实践指南(适用于非孕妇或免疫功能低下成人)
- 美国传染病学会发布的《婴幼儿、儿童、青少年和成人免疫接种计划:临床实践指南》
- SHEA/IDSA 实施抗菌药物管理计划的临床实践指南
- IDSA 2017 年感染性腹泻诊断和治疗临床实践指南
- IDSA 2018 年季节性流感诊断、治疗、化学预防和机构暴发管理临床实践指南更新
- IDSA 2024 年指南更新:成人、儿童和孕妇复杂性腹腔内感染的风险评估、影像诊断和微生物学评估
- 对免疫状况不明或未完成的人员进行疫苗接种:自2026年2月起
- 美国精神卫生与临床优化研究所/美国传染病学会利什曼病诊断和治疗临床实践指南