
Contents
Guideline for Elemental Impurities
Step 5
2014/12/16
目录
- 前言
- 范围
- 潜在元素杂质的安全评估
- 3.1 口服、注射和吸入给药中元素杂质安全评估的原则
- 3.2 其它给药途径
- 3.3 元素杂质水平高于已建立的PDE时的论证
- 3.4 注射用药
- 元素分类
- 元素杂质的风险评估和控制
- 5.1 通用原则
- 5.2 元素杂质的潜在来源
- 5.3 潜在元素杂质的识别
- 5.4 建议中风险评估中考虑的元素
- 5.5 评估
- 5.6 风险评估过程总结
- 5.7 生物技术衍生产品特殊考虑
- 元素杂质的控制
- PDE和关注限度之间的转换
- 物种形成和其它考虑
- 分析方法
- 生命周期管理
术语
参考文献
附录1:建立暴露限度的方法
附录2:已建立的元素杂质PDE
附录3:单独安全评估
附录4:举例说明
1. 介绍
药品中的元素杂质可能会有几个来源,它们可能是有意加入合成反应的催化剂的残留,也可能是作为杂质出现(例如,通过与工艺设备或容器/密闭系统相互反应,或出现在药品的组分中)。由于元素杂质并不给患者提供任何治疗益处,其在药品中的水平应被控制在可接受限度以内。本指南分为三个部分:潜在元素杂质毒性数据的评估、为每个毒性关注元素建立PDE 值,以及应用基于风险的方法来控制药品中的元素杂质。如果药品中的元素杂质没有超过PDE 阈值的话,申报人不需要根据其工艺能力加严限度。本指南中建立的PDE 阈值足以保护所有患者人群的公共健康。在有些情况下,如果毒性阈值以下的元素杂质水平表示出对药品的其它质量属性有影响(例如,对药品降解有催化作用的元素),则可能需要保证一个更低的元素杂质水平。另外,对于具有较高PDE 值的元素,可能需要从药品质量的角度,以及要参照的其它指南(例如ICH Q3A)来考虑其它限度。
本指南给出一个采用ICH Q9 中所述风险管理原则来评估和控制药品中元素杂质的方法。该方法提供了一个基于风险控制策略的平台来限制药品中的元素杂质。
2. 范围
本指南适用于新的制剂产品(如ICH Q6A 和Q6B 定义)和含有已有原料药的新药品。含有纯化后的蛋白质和多肽(包括采用复合或非复合来源生产的蛋白质和多肽)的药品、其衍生物,以及其复方药品(例如,偶合物)在本指南适用范围内。含有合成多肽、多核苷酸和低聚糖的药品也适用本指南。
本指南不适用于草药产品、放射性药品、疫苗、细胞代谢物、DNA 产品、过敏提取物、细胞、全血、细胞血成分或血液制品,包括血浆和血浆制品、非系统循环用透析液,和用于治疗用途加入的元素。本指南不适用于基于基因(基因治疗)、细胞(细胞治疗)和组织(组织工程)的药品。在有些地区,这些产品是作为先进治疗药品的。
本指南不适用于研发的临床研究阶段药品。由于商业过程是在不断发展的,评估新药中可能出现的元素杂质时也可应用本指南中的原则。
在本指南由ICH 发布后36个月内,不需要对已有产品应用Q3D。
3. 潜在元素杂质的安全评估
3.1 口服、注射和吸入给药途径的元素杂质安全评估原则
用于建立各元素杂质的方法在附录1 中进行了讨论。在本指南中评估的元素,是通过对科学杂质、政府研发报告和研究、国际法规标准(适用于药品)和指南、以及法规当局研究和评估报告里公众可以获得的数据进行审核得到的。该方法是根据ICH Q3C:残留溶剂中所述的原则制订的。对可以获得的资料进行审核以建立口服、注射和吸入PDE 值。为了实用,附录2 里表A.2.1 中适用于药品的PDE 阈值被修约至1 位或2 位有效数字。
附录3 包括了一份各元素PDE 设定的关键研究识别安全评估总结。对于铱、锇、铑和铷没有足够的数据设定口服给药途径的PDE 阈值。这些元素的PDE 值是基于其与钯的相似性上建立的。
在建立PDE 所进行的安全评估中考虑的因素按大致的相关性顺序列出如下:
- 药品中的元素可能的氧化状态
- 当其提供可用信息时,人类暴露量和安全数据
- 最相关的动物研究
- 给药途径
- 相关终点
Standards for daily intake for some of the elemental impurities discussed in this guideline exist for food, water, air, and occupational exposure. Where appropriate, these standards were considered in the safety assessment and establishment of the PDEs.
在本指南中讨论的有些元素杂质日服用量的标准…
一般使用最长的动物研究时长来建立PDE 值。如果有一个较短的动物研究时长被认为是最为相关的,则在单个安全评估中给出了其理由。
相对使用粒子的研究,使用可溶性盐(可获得时)进行的吸入研究优先用于吸入安全性评估和计算吸入PDE 值。根据可获得的数据,吸入PDE 值是基于局部(喷雾系统)或系统性毒性的。对于为了吸入给药建立的PDE 值(适用时,和口服或注射途径),剂量一般统一为24 小时7 天暴露时长。
如果没有数据,和/或有数据但认为不足以用于注射和/或吸入途径的安全评估,则基于口服生物利用度的修正因子用于从口服PDE 来推导PDE:
- 口服生物利用度 <1%: 除以100 作为修正因子;
- 口服生物利用度 ≥ 1% and <50%: 除以10 作为修正因子;
- 口服生物利用度 ≥50% and <90%: 除以2 作为修正因子;以及
- 口服生物利用度 ≥ 90%: 除以1 作为修正因子。
如果没有口服生物利用度数据或职业吸入暴露限,则在根据口服PDE值计算出PDE值后除以修正因子100(参考文献1)。
3.2 其它摄入途径
PDE 是针对口服、注射和吸入给药途径建立的。如果需要其它给药途径的PDE,则可以使用本指南的概念来推导PDE。评估结果可能会升高或降低已建立的PDE 值。从一种给药途径推导出另一种给药途径的PDE 值的计算过程包括以下:
- 将附录3 中的口服PDE 作为建立摄入途径特定PDE 的起始点。基于科学评价,注射和吸
入PDE 可能是一个更适当的起始点。 - 评估该元素杂质在通过预定给药途径摄入时是否预期产生局部影响
- 如果预期有局部影响,需要评估是否要对已建立的 PDE 进行修正
- 考虑预期产生这些影响时的剂量/暴露量,与用于设定已建立的PDE 所用的不良反应相比较
- 如果预期没有局部影响,则对于已建立的 PDE 不需要进行调整
- 如果可以,应评估该元素的通过预定给药途径的生物利用度,并将此与该元素通过已建立PDE 的给药途径的生物利用度进行比较
- 如果观察到差异,则可能需要对已建立的PDE 值使用校正因子。例如,如果预期有局部影响,如果一种元素的口服生物利用度为50%,一种元素的生物利用度在预定的给药途径为10%,则可以使用5作为校正因子
- 如果提议一种新的给药途径的 PDE 相对于已建立的PDE 有增加,则可能需要考虑对质量属性
3.3 元素杂质水平高于已建立的PDE水平时的论证
元素杂质水平高于已建立的PDE 时(参见表A.2.1),在特定情况下可能也可以接受。这些情况可能包括但不仅限于以下情形:
- 间歇给药
- 短期给药(即,30 天或更短)
- 特定指示(例如,生命威胁、药品供给不足、罕见病)
以下提供了使用修正因子的子因子方法(参见2.3)对升高的元素杂质水平进行论证的例子。其它方法也可以用来论证较高的杂质水平。提议任何高于已建立的PDE 的杂质水平均需根据各案进行论证。
例1:元素X 出现在口服药品中。元素X 各论见附录3,其NOAEL 水平为1.1mg/kg/天。修正因子F1-F5 分别设定为5、10、5、1 和1。使用附录1 中所述的修正因子标准方法,PDE 计算如下:
PDE = 1.1 mg/kg/d × 50 kg / 5 × 10 × 5 × 1 × 1 = 220 μg/day
修正因子F2(默认=10)可以分成2 个子因子,一个作为毒性动力学(TK),另一个作为毒理动力学,2 个因子范围均为1-3.16。采用5 天使用血浆半衰期,对于每次一周摄入(-1半衰期)TK 调整因子可以降低为1.58,对于每次一个月摄入(-5 个半衰期)可以降低为1。通过对F2 使用子因子方法,元素X 每次一周摄入建议水平可以计算如下:
建议水平 = 1.1 mg/kg/d × 50 kg / 5 × (1.6 × 3.16) × 5 × 1 × 1 = 440 μg/day
为了实用,该值修约为400μg/day。
例2:TK 调整因子方法可能也适用于未采用修正因子方法建立的元素杂质。对于元素Z,最低风险水平(MRL)为0.02mg/kg/天用以计算口服PDE 值。从文献来看,血浆半衰期报道为4 天。该元素是口服给药中的一个杂质,药品每三周给药一次(-5 个半衰期)。使用一级动力学,已建立的PDE 为1000μg/天修正如下:
建议水平 = 0.02 mg/kg/d × 50 kg / 1/3.16 = 3.16 mg/day
为实用起见,该值修约为3000μg/天。
3.4 注射用药
注射用药如果最大日给药体积达到2L,则可以使用最大日给药体积来计算PDE的允许浓度。对于日剂量在标签上注明和/或临床确定的药品,可以超过2L(例如,生理盐水、葡萄糖、总注射用营养、冲注洗剂),2L 的体积可以用于计算PDE 的允许浓度(参考文献4)。
4. 元素分类
本指南中包括的元素已根据其毒性(PDE)及在药品中出现的可能性分入三类。出现可能性是从几个因素中推导出的,包括:在制药工艺中使用的可能性、制药工艺中使用的原料里含有的杂质会产生共析的杂质可能性,以及观察到自然中富含的元素和在环境中广泛分布的元素。根据本指南的目的,一种在自然中存量较低的元素指其自然含量<1个原子/106 个硅原子(参考文献5)。分类表目的是将风险评估的焦点集中在那些最毒,且最可能出现在药品中的元素上(参见表5.1)。元素杂质分类为:
第1类:元素砷、镉、汞和铅是对人有毒性的物质,已限制或不再用于药品生产中。其在药品中出现一般是来自于通常使用的物料(例如,矿物质辅料)。由于其独特的属性,这四种元素需要在风险评估中进行评价,要针对元素杂质的所有潜在来源以及所有的摄入途径。风险评估的结果将决定这些组成是否需要增加控制,在有些情况下要包括对一类元素的检测。不需要对所有成分进行一类元素杂质的检测,只有在风险评估认为需要对其进行适当控制以保证符合PDE 要求时才要进行检测。
第2类:本类别中的元素一般被认为是与摄入途径相关的人类有毒物质。根据其出现在药品的相对可能性,2 类元素又被分为2A 和2B 两个子类。
- 2A 类:在药品中出现可能性相对较高的元素,因而需要对所有元素杂质的潜在来源及所有摄入途径(如所指)进行风险评估。2A 类元素为钴、镍和钒。
- 2B 类:由于自然含量较低、与其它物料共存可能性较低,在药品中出现的可能性较低的元素。因此,除非其在原料药、辅料或药品的其它成分生产中被有意加入,否则可能被排除在风险评估以外。2B 类的元素杂质包括:银、金、铱、锇、钯、铂、铑、铷、硒和铊。
第3类:本类的中元素在口服摄入时具有相对较低的毒性(高PDE,通常>500 μg/day),但可能在吸入和注射给药的风险评估中需要进行考虑。对于口服摄入,除非这些元素被有意加入,否则不需要在风险评估中进行考虑。对于注射和吸入给药药品,除非给药途径的PDE超过500μg/day,否则在风险评估中要评价这些元素杂质出现的可能性。本类中的元素包括钡、铬、铜、锂、钼、锑和锡。
其它元素:有些元素杂质因为其较低的毒性和/或在地方法规中的要求不同,其PDE 还没有建立,在本指南中并未说明。如果这些元素杂质出现或包括在药品中,其它指南和/或地方性法规和规范可能适用于特殊的元素(例如,铝,损害肾功能,锰和锌对于肝功能不全的病人),或对药品成品的质量考虑(例如,钨杂质在治疗性蛋白质中出现)。这些特殊考虑的元素包括:铝、硼、钙、铁、钾、镁、锰、钠、钨和锌。
5. 元素杂质的风险评估和控制
在建立药品中元素杂质的控制方式时,要考虑ICH Q9 中所述的质量风险管理的原则。风险评估应基于科学知识和原则,应将对产品和其生产工艺的了解(ICH Q8 和Q11)与对患者的安全考虑相关联。对于元素杂质来说,药品风险分析就应聚焦于结合本指南中所给出的PDE 来评估一种药品中的元素杂质水平。风险评估的资料包括,但不仅限于:申请人产生的数据、原料药和/或辅料生产商提供的资料,和/或在公开的文献中可以获得的数据。
申报者应以适当的方式记录风险评估和控制方法。风险评估和努力水平和正式程度应与风险水平相称。没有必要每次都使用正式的风险管理过程(使用已知的工具和/或正式程序,例如,标准操作程序)。也可以使用非正式的风险评估过程(使用经验工具和/或内部程序)。
风险评估中辅助工具在ICH Q8 和Q9 中已有描述,本指南中不再赘述。
5.1 通则
出于本指南的目的,风险评估过程可以描述为以下三步:
- 识别已知和潜在可能进入药品的元素杂质来源,
- 通过测试已知或预期杂质,将其水平与已有PDE 值比较,评估药品中特殊的元素杂质出现的可能性
- 总结和记录风险评估。识别出工艺中嵌入控制是否充分,或识别出要考虑增加控制来限制药品中的元素杂质
在很多情况下,这些步骤其实是同步的。风险评估的结果,可以是一个迭代的结果,用以建立一种方法来保证潜在元素杂质不超过PDE 值。
5.2 元素杂质的潜在来源
在考虑一种药品的生产时,元素杂质的潜在来源有很多。
- 在生产原料药、辅料或其它药品成分时有意加入的元素的残留杂质(例如催化剂)。原料药的风险评估要说明元素杂质出现在药品中的可能性
- 非有意加入,可能会在药品制备过程中出现在原料药、水或辅料中的元素杂质
- 可能从生产设备引入原料药和/或制剂的元素杂质
- 可能从容器密闭系统中溶出至原料药和制剂的元素杂质
下图表示了一种药品生产中所用的典型物料、设备和成分的一个例子。通过单独或上列潜在来源的联合,每种来源均可能引起药品中的元素杂质污染。在风险评估中,任何一种来源的潜在作用均应进行考虑,以确定对药品造成的总体元素杂质污染。
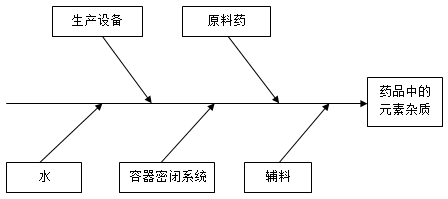
* 通过对工艺的了解、设备的选择、设备确认和GMP,可以降低元素杂质引入风险。
** 如果在生产工艺中使用了纯化水或注射用水,从水中引入元素杂质的风险可能通过符合药典水质量来降低(例如,欧洲药典、日本药典、美国药典)。
5.3 潜在元素杂质的识别
来自有意加入的催化剂和无机试剂的潜在元素杂质:如果有意地加入了表5.1 中的任何元素,则应在风险评估中考虑。对此类情况,潜在杂质是已知的,控制元素杂质的技术易于制订。
可能会出现在原料药和/或辅料中的潜在元素杂质:在非有意加入情况下,有些元素杂质可能会出在有些原料药和/或辅料中。在风险评估中要反映药品中含有这些元素的可能性。
对于口服给药途径,风险评估应评价药品中含有1 类和2A 类元素杂质的可能性。对于注射和吸入给药途径,风险评估应评价含有1 类、2A 类和3 类元素杂质的可能性,如表5.1 所示。
生产设备中生成的潜在元素杂质:从该途径来源的元素杂质可能会比较有限,在风险评估中需要包括的元素杂质种类将取决于药品生产所用的生产设备。工艺知识申报、设备选择、设备确认和GMP 控制能保证生产设备来源的元素杂质在一个较低的水平。应根据与药品成分接触的生产设备的部件成分知识,对特别关注的元素杂质进行评估。该类来源的元素杂质风险评估可以用于使用类似设备链和工艺的多个药品。
一般来说,在评估从生产设备清除或析出元素杂质可能性时,用于制备指定原料药的工艺条件要比药品制备工艺条件严苛的多。来自制剂工艺设备中的元素杂质一般预期会低于来自原料药工艺设备的杂质。但是,如果根据工艺知识或理解并不是这样的话,则申报人应在风险评估中考虑来自制剂生产设备的元素杂质结合的可能性(例如,热融挤压)
从容器密闭系统中溶出的元素杂质:对可能从容器密闭系统引入的潜在元素杂质的识别应基于特殊药品类型和其它包装间的可能的相互反应的科学理解。如果对结构材料的审核证明容器密闭系统不含有任何元素杂质,则不需要进行额外的风险评估。我们认识到元素会溶出至固体剂型的可能性是非常小的,不需要进在风险评估中进行深入考虑。对于液体和半固体剂型,则在药品的货架期内,元素杂质会从容器密闭系统中溶出到药品中的可能性会比较大,此时应对容器密闭系统的潜在溶出物质进行研究(在清洁、灭菌、辐射后等)。该类元素杂质一般在药品的容器密闭系统的评估中要重点论述。
要考虑的因素(对于液体或半固体剂型)包括但不仅限于:
- 亲水性/吸湿性
- 离子含量
- pH;
- 温度(冷链 VS 室温和处理条件)
- 接触面积
- 容器/组件成分
- 最终灭菌
- 包装过程
- 部件灭菌
- 存贮时长
5.4 风险评估中要考虑的要素建议
以下表格对于风险评估中需要包括的元素杂质做出了建议。该表可以用于药品中所有来源的元素杂质。
表5.1: 在风险评估中要考虑的元素
| 元素 | 分类 | 如果有意加入 | 如果无意加入 | ||
|---|---|---|---|---|---|
| – | – | 所有给药途径 | 口服 | 注射 | 吸入 |
| Cd 镉 | 1 | Yes | Yes | Yes | Yes |
| Pb 铅 | 1 | Yes | Yes | Yes | Yes |
| As 砷 | 1 | Yes | Yes | Yes | Yes |
| Hg 汞 | 1 | Yes | Yes | Yes | Yes |
| Co 钴 | 2A | Yes | Yes | Yes | Yes |
| V 钒 | 2A | Yes | Yes | Yes | Yes |
| Ni 镍 | 2A | Yes | Yes | Yes | Yes |
| Tl 铊 | 2B | Yes | No | No | No |
| Au 金 | 2B | Yes | No | No | No |
| Pd 钯 | 2B | Yes | No | No | No |
| Ir 铱 | 2B | Yes | No | No | No |
| Os 锇 | 2B | Yes | No | No | No |
| Rh 铑 | 2B | Yes | No | No | No |
| Ru 铷 | 2B | Yes | No | No | No |
| Se 硒 | 2B | Yes | No | No | No |
| Ag 银 | 2B | Yes | No | No | No |
| Pt 铂 | 2B | Yes | No | No | No |
| Li 锂 | 3 | Yes | No | Yes | Yes |
| Sb 锑 | 3 | Yes | No | Yes | Yes |
| Ba 钡 | 3 | Yes | No | No | Yes |
| Mo 钼 | 3 | Yes | No | No | Yes |
| Cu 铜 | 3 | Yes | No | Yes | Yes |
| Sn 锡 | 3 | Yes | No | No | Yes |
| Cr 铬 | 3 | Yes | No | No | Yes |
5.5 评估
在对潜在元素杂质进行识别后,可能会有两种结论:
1) 风险评估未能识别出任何潜在元素杂质。风险评估的结论和支持性资料和数据要进行记
录。
2) 风险评估识别出了一个或多个潜在元素杂质。对所有识别出的元素杂质,如果该元素杂
质有多个来源,要考虑进行风险评估。并记录评估的结论和支持性资料。
申报人的风险评估可以利用原料药、辅料、容器密闭系统和生产设备供应商提供的关于潜在元素杂质的信息。支持风险评估的数据可以来自于许多来源,包括但不仅限于:
- 之前的知识
- 出版的文献
- 从类似工艺中产生的数据
- 供应商的信息或数据
- 药品组分检测
- 药品检测
在风险评估中,很多因素会对药品中潜在杂质的水平造成影响,因此也需要在风险评估中进行考虑。这包括但不仅限于:
- 进一步加工中除去元素杂质的有效性
- 自然富含的元素(特别重要的是没有添加单身的杂质类别)
- 特定来源的元素杂质浓度已有知识
- 药品组分
5.6 风险评估过程的总结
通过对相关产品或组成相关数据,结合在产品或工艺中获得的信息和知识进行审核,进行风险评估,以识别明显可能在药品中观察到的元素杂质。
总结要考虑相对于元素杂质PDE 值,已观察到的或预期的元素杂质水平的显著性。这里,将药品中已建立的PDE 值的30%定义为控制阈值,作为对已观察到的元素杂质水平的显著性的衡量方法。
如果药品中所有来源的总元素杂质水平预期会保持一致,且低于PDE 值的30%水平,而申报人已对数据进行了适当的评估,证明对元素杂质的控制已经足够充分,则不需要采用更多的控制措施。
如果风险评估未能证明一种元素杂质的水平具有一致性且低于控制阈值,则要建立控制措施来保证元素杂质水平不会超过药品的PDE 值(参见第6 部分)。
元素杂质水平的可变性应分解到药品阈值控制的应用中去。可变性的来源可能包括:
- 分析方法的可变性
- 特定来源中的元素杂质的可变性
- 药品中元素杂质水平的可变性
在提交申报时,如果没有其它论证,一种元素杂质的水平和可变性可以通过提供成分或药品生产的3 批生产规模或6 批中试规模具代表性的批次数据来建立。对于有些具有内在可变性的成分(例如,矿物质辅料),在应用控制阈值时可能需要额外的数据。
许多总结和记录风险评估的方法都是可以接受的,包括:表格、所有考虑因素的书面总结、评估的结论。总结应识别出元素杂质、其来源、以及控制方式,需要时要制订可接受标准。
5.7 生物技术衍生药品的特殊考虑
对于生物技术衍生药品,在原料药阶段引起安全关注的元素杂质的风险被认为是很低的。这很大程度是因为1)元素不是在生物技术药品生产中典型用作催化剂或试剂,2)在细胞发酵过程中培养基补料时加入的元素为痕量水平,不会累积,在进一步加工时会被显著稀释/清除,3)在生物技术生产中使用的典型的纯化过程,如萃取、色谱分离和透析或超滤-渗滤(UF/DF),具备能力将细胞生长/发酵步骤或与生产设备接触过程中引入的元素清除至可忽略的水平。在这种情况下,对生物技术原料药中元素杂质的特别控制通常并不需要。如果生物技术衍生原料药含有合成结构(例如抗体药偶合物),需要评估是否需要对小分子成分中的元素杂质进行适当的控制。
6. 元素杂质的控制
控制元素杂质是药品全面控制策略的一部分,它能保证元素杂质不超过PDE 值。当元素杂质有可能超过控制阈值时,需采取更多措施来保证其水平不会超过PDE 值。申报人可以采用的措施包括但不仅限于:
- 改进生产工艺步骤,通过特定或非特定的精制步骤将元素杂质降低至控制阈值以下
- 实施中控或上游控制,用以将元素杂质的浓度限定在制剂的控制阈值以下
- 建立辅料或原料的质标准限度(例如,合成中间体)
- 建立原料药质量标准限度
- 建立制剂质量标准限度
- 选择适当的容器包装系统
根据ICH Q6A 中所述的原则,元素杂质可能需要定期进行测试。需要包括在法规申报中的
元素杂质控制资料包括,但不仅限于,风险评估总结,必要时提交适当的数据,以及已建立
的用于限制元素杂质的控制方法描述。
7. 将PDE值和浓度限度互相转换
PDE 值以微克每天表示(μg/天),在本文中给出的是可能包括在药品最大日服用量里的每个元素允许的最大数量。由于PDE 反映的只是来自药品的总暴露量,因此需要将PDE 值转换为浓度,作为工具用于评估药品或其成分中的元素杂质。本部分列出的方法描述了一些可接受的方法,用于建立药品或其成分中的元素杂质浓度,从而保证药品不会超过PDE 值。只要所导出的允许浓度能保证药品不会超出PDE 值,申报人可以选择这些方法中的任何一种。在选择特定的方法时,申报人必须知晓,或假定,药品的日摄入量。允许浓度限度可以用于:
- 作为风险评估的工具,与观察到或预期的 PDE 水平进行比较
- 在与供应商的讨论中帮助建立上游控制可以保证产品不会超过 PDE 值
- 在研究元素杂质中控时建立目标浓度
- 在法规申报中传递元素杂质控制的信息
正如第5.2 部分所讨论的,药品中的元素杂质有多种来源。在使用下述任何一种谅埋,在计算含有杂质的组分中(辅料和原料药)的最大允许浓度时,要考虑来自容器密闭系统和生产设备的元素杂质。如果在风险评估中已确定容器密闭系统和生产设备不会增加药品中的元素杂质,则不需要考虑。如果容器密闭系统和生产设备存在可能引入元素杂质,则在计算辅料和原料药中允许浓度前,可以从PDE 中减去这些来源的估计的日摄入量。
方法1:日摄入量不超过10g的药品的药品组分中元素通用允许浓度限度
本方法无意暗示所有元素均以相同浓度出现,只是提供了一种简化的方法来进行计算。
本方法假定药品的日摄入(量)为10g 或更低,风险评估中识别的元素杂质(目标元素)出现在药品的所有成分中。使用以下公式1,药品的日摄入量为10g,本方法计算出药品中每种成分的允许目标元素浓度。本方法,对于每一目标元素,允许对于每一成分制订固定的通用最大浓度,单位为mg/g。附录2 表A.2.2 中给出了允许浓度。
如果药品中的所有成分均不超过方法2a 中在风险评估中已识别的所有目标元素的浓度,则所有这些成分均可以以任何比例用于药品生产。本方法例子在附录4 表A.4.2 中。如果附录2 表A.2.2 中的允许浓度不适用,则应使用方法2a、2b 或3 的数据。
方法2a:具有特定日摄入量的药品中药品组分的通用允许浓度限度
本方法与方法1 类似,除了药品日摄入量不会假定为10 克。每种元素的通用允许浓度采用公式1 和实际最大日摄入量来计算。
本方法,对于每一目标元素,允许根据所提供的实际日摄入量,确定每种成分的固定通用的最大浓度(单位为mg/g)。本方法例子在附录4 表A.4.3 中。
如果药品中的所有成分均不超过方法2a 中在风险评估中已识别的所有目标元素的浓度,则所有这些成分均可以以任何比例用于药品生产。
方法2b:具有特定的日摄入量的药品的单个成分中元素的允许浓度限度
本方法要求有另外的资料,申报人可以根据特定药品成分中出现的特定元素杂质的可能性对数据进行整合。申报人可以根据组分中元素的分布来设定允许浓度(例如,所讨论的元素出现在组分中浓度较高)。对于每种被识别为药品组分中可能出现的元素,药品成品中元素杂质的最高期望质量可以通过将每种组分物料的质量乘以申请人建立的每种物料中的允许浓度,然后将药品中所有成分中的元素杂质质量相加来计算,如公式2 所述。除非根据本指南其它相关部分进行了论证,否则药品里元素杂质的总质量应符合附录2 表A.2.1 中给定的PDE 值。如果风险评估已认定某特定组成中特定的元素不是潜在杂质,则不需要为该元素在该组分中的含量建立定量结果。本方法允许药品中特定组分中某元素的最高允许浓度高于方法1 或方法2a 中的限度,但这就表示药品中其它组成中的允许浓度要更低。公式2 可以用来证明通过控制各组分里特定组分中每种元素的限度,最终保证药品符合PDE 要求。

K = 药品中N组成的序号
Ck = 组分K中元素杂质的允许浓度(μg/g)
Mk = 药品最大日摄入量中组分K 的质量(g)
本方法的例子参见附录4 表A.4.4-A.4.5。
方法3:制剂分析
各元素的浓度可以在制剂成品中检测。公式1 可以和总最大日剂量一起用于计算允许的最
高元素杂质浓度。本方法例子参见附录4,表A.4.6.
8. 元素形态和其它考虑
元素形态是指化学形态中元素的分布情况,包括同位素组成、电子或氧化状态和/或复合或分子结构。如果同一元素的不同形态为已知,则PDE 使用预期会出现在药品中的化学形态的毒性资料来计算。
如果元素杂质的测定被用于风险评估,药品中总元素杂质水平可以用于评估是否符合PDE要求。申报人不需要提交物种资料,但是,当所识别的物种比附录3 所用物种获得的毒性更高或更低时,这些资料可以用于论述较低的或较高的杂质水平。
如果在风险评估中使用了所有组分中总元素杂质水平,则申报者不需要提交单个组分中所发现的元素杂质的放行资料。但是,该资料可以用于论述单个组分中元素杂质水平高于药品中总元素杂质水平的情况。
9. 分析方法
元素杂质的检测应使用适当的分析方法,适用于其既定目的。除另有论证外,在风险评估中识别出需要控制的每种元素杂质均需有特定的检测方法。可以使用药典方法或适当的替代性方法来测定元素杂质的水平。
10. 生命周期管理
在ICH Q10 中描述了质量体系和管理责任,其目的是鼓励在生命周期各阶段使用基于科学和基于风险的方法,从而促进整个产品的生命周期内的持续改进。产品和工艺知识管理应从研发阶段开始,贯穿整个商业历程,直至产品退市。
从研发阶段获得的知识,与商业生产经验和数据相结合,可以用于进一步改进工艺知识和工艺性能。这种改进可以增强对元素杂质的控制。我们认识到从有些组分中获得的元素杂质数据在本指南公布时仍是很有限的,这可能要求申报者建立特定的控制。如果在后期研究获得更多数据,可能会导致对控制方式的修改。
如果对药品或组成的变更可能对药品中元素杂质的含量产生影响,则应重新进行风险评估,包括对已建立的元素杂质控制方式的评估。这类变更可能包括但不仅限于:合成路线变更、辅料供应商变更、原料变更、工艺变更、设备变更、容器密闭系统或设施变更。所有变更均应通过内部变更管理流程进行管理,必要时应满足地区法规要求。
术语(未译)
参考文献
- Ball D, Blanchard J, Jacobson-Kram D, McClellan R, McGovern T, Norwood DL et al. Development of safety qualification thresholds and their use in orally inhaled and nasal drug product evaluation. Toxicol Sci 2007;97(2):226-36.
- IPCS. Principles and methods for the risk assessment of chemicals in food, chapter 5: dose-response assessment and derivation of health based guidance values. Environmental Health Criteria 240. International Programme on Chemical Safety. World Health Organization, Geneva. 2009;Table 5.5.
- US EPA. 0410 Boron and Compounds. Integrated Risk Management System (IRIS).
2004. - Holliday MA, Segar WE. The maintenance need for water in parenteral fluid therapy. Pediatrics 1957;19:823-32.
- Haxel GB, Hedrick JB, Orris GJ. Rare earth elements-critical resources for high technology. US Geological Survey 2005;Fact Sheet 087-02.
附录1:建立暴露限度的方法
对于大多数元素,本指南中的元素杂质可接受暴露水平是根据在药品中设定暴露限度的方法(参考文献1),通过PDE 值的计算来建立的,以及被国际化学安全组织(IPCS)用于评估人体健康化学风险(参考文献2)的方法。这些方法类似于美国环境保护署(USEPA)一体化风险信息系统,美国FDA(参考文献3)和其它组织使用的方法。这里列出的方法是为了对PDE 值的来源有更好的理解。当MRL 被用于建立PDE 值时,不需要使用另外的修正因子,因为这些修正因子已被整合在了MRL 的计算中。对于致癌元素,单位风险因子被用于建立PDE 值,其中使用1:100000 风险水平,这些在附录3 各论中进行了描述。有些吸入途径的PDE 是采用了职业暴露限度来计算的,使用了修正因子,考虑了对吸入系统的所有特殊影响。
采用最相关的动物研究所获得的NO(A)EL 或LO(A)EL 计算PDE 如下:
PDE = NO(A)EL x Mass Adjustment/[F1 x F2 x F3 x F4 x F5] (A.1.1)
PDE 最好是从NO(A)EL 计算出。如果没有NO(A)EL 值,也可以使用LO(A)EL 值。对于将数据联系到人类,建议使用修正因子,该因子类似于环境健康标准(参考文献2)中使用的“不确定因子”,以及药典论坛中使用的“修正因子”或“安全因子”。
修正因子如下:
F1=不同物种间外推时所用的修正因子
F1=1,人体数据
F1=5,从大鼠外推至人类
F1=12,从小鼠外推至人类
F1=2,从狗外推至人类
F1=2.5,从家兔外推至人类
F1=3,从猴子外推至人类
F1=10,从其它动物外推至人类
F1 考虑了可比表面积:与物种和人有关的体重比例。表面积计算如下:
S = kM0.67 (A.1.2)
其中:M=体重,常数K 取为10.用于公式A.1.2 中的体重是表A.1.1 中给出的数据。
F2=不同各体间的差异考虑,取为10
所有元素杂质通常将该因子取为10,在本指南中一律取10
F3=短期暴露毒性研究中考虑的变动因子
F3=1,持续至少半个生命周期(啮齿动物或家兔为1 年,猫、狗和猴子为7 年)的研究
F3=1,覆盖整个器官形成阶段的生殖研究
F3=2,对啮齿动物持续6 个月的研究,或对非啮齿动物持续3.5年的研究
F3=5,对啮齿动物持续3 个月的研究,或对非啮齿动物持续2年的研究
F3=10,更短期的研究
在所有情况下,如果研究时长在两个因子中间,则采用更大的因子,例如,对啮齿动物持续9个月的研究取修正因子为2
F4=有严重毒性时的因子,例如,非基因毒性致癌性,神经毒性或致畸性。在生殖毒性研究中,可以采用以下因子:
F4=1,致命毒性伴随母体毒性
F4=5,致命毒性,无母体毒性
F4=5,致畸性影响,伴随母体毒性
F4=10,致畸性影响,无母体毒性
F5=没有NOEL 值时的变动因子
F5=1,使用NOEL
F5=1-5,使用NOAEL
F5=5-10,使用LOEL
F5=10,使用观察到不良反应的最低水平(LOAEL)
对于大多数元素,NOAEL 被用于设定口服PDE 值,当研究未调查NOALE 和NOEL 之间的差异时,所选择的用于确定PDE 的剂量毒性不被作为“副反应”时,使用F5=1。
体重调整是假定男女性成人体重均为50kg。该相对较低的体重相比于常用的60kg 或70kg体重值可以提供更多的保护因素。我们认识到有些病人的体重低于50kg,这些病人被认为需要使用内在安全因子来确定PDE 值,这在生命周期研究中常会用到。对于铅,儿科人群会被认为是最易感人群,从该人群中获得的数据被用于建立PDE 值。因此,该PDE 值被认为适用于儿科药。
举例说明公式A.1.1 的应用,考虑TVERMOES(参考文献4)中总结的钴人体自愿者的毒性研究数据,红血球增多症的NOALE 值定为1mg/天,本研究中钴的PDE 值计算如下:
PDE = 1 mg/day /[1 x 10 x 2 x 1 x 1] = 0.05 mg/day= 50 μg/day
在本例中
F1=1,对人体的研究
F2=10,考虑个体间差异
F3=2,因为研究时长为90 天
F4=1,因为没有遇到严重毒性
F5=1,因为使用了NOALE
表A.1.1:在本文件中用于计算的取值
| 大鼠体重 | 425g | 小鼠吸入体积 | 43L/day |
|---|---|---|---|
| 怀孕大鼠体重 | 330g | 家兔吸入体积 | 1440L/day |
| 小鼠体重 | 28g | 豚鼠吸入体积 | 430L/day |
| 怀孕小鼠体重 | 30g | 人体吸入体积 | 28,800L/day |
| 豚鼠体重 | 500g | 狗吸入体积 | 9,000L/day |
| 恒河猴体重 | 2.5kg | 猴子吸入体积 | 1,150L/day |
| 家兔体重(怀孕或未孕) | 4kg | 小鼠消耗水量 | 5mL/day |
| 比格犬体重 | 11.5kg | 大鼠消耗水量 | 30mL/day |
| 大鼠吸入体积 | 290L/day | 大鼠消耗食物量 | 30g/day |
参考文献
- United States Pharmacopeial Convention, Pharmacopeial Forum, Nov-Dec 1989.
- IPCS. Assessing Human Health Risks of Chemicals: Derivation of Guidance Values for Health-based Exposure Limits, Environmental Health Criteria 170. International Programme on Chemical Safety. World Health Organization, Geneva. 1994.
- US FDA, Guidance for Industry and Other Stakeholders: Toxicological Principles for the Safety Assessment of Food Ingredients (Redbook 2000), available at http://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/IngredientsAdditivesGRASPackaging/ucm2006826.htm.
- Tvermoes BE, Unice KM, Paustenbach DJ, Finley BL, Otani JM, Galbraith DA. Effects and blood concentrations of cobalt after ingestion of 1 mg/d by human volunteers for 90 d. Am J Clin Nutr 2014;99:632-46.
附录2:已建立的元素杂质PDE
表A.2.1: 元素杂质允许日暴露量1
| 元素 | 分类2 | 口服PDE | 注射PDE | 吸入PDE |
|---|---|---|---|---|
| Cd 镉 | 1 | 5 | 2 | 2 |
| Pb 铅 | 1 | 5 | 5 | 5 |
| As 砷 | 1 | 15 | 15 | 2 |
| Hg 汞 | 1 | 30 | 3 | 1 |
| Co 钴 | 2A | 50 | 5 | 3 |
| V 钒 | 2A | 100 | 10 | 1 |
| Ni 镍 | 2A | 200 | 20 | 5 |
| Tl 铊 | 2B | 8 | 8 | 8 |
| Au 金 | 2B | 100 | 100 | 1 |
| Pd 钯 | 2B | 100 | 10 | 1 |
| Ir 铱 | 2B | 100 | 10 | 1 |
| Os 锇 | 2B | 100 | 10 | 1 |
| Rh 铑 | 2B | 100 | 10 | 1 |
| Ru 铷 | 2B | 100 | 10 | 1 |
| Se 硒 | 2B | 150 | 80 | 130 |
| Ag 银 | 2B | 150 | 10 | 7 |
| Pt 铂 | 2B | 100 | 10 | 1 |
| Li 锂 | 3 | 550 | 250 | 25 |
| Sb 锑 | 3 | 1200 | 90 | 20 |
| Ba 钡 | 3 | 1400 | 700 | 300 |
| Mo 钼 | 3 | 3000 | 1500 | 10 |
| Cu 铜 | 3 | 3000 | 300 | 30 |
| Sn 锡 | 3 | 6000 | 600 | 60 |
| Cr 铬 | 3 | 11000 | 1100 | 3 |
1 本表中报告的PDE(μg/day)是基于附录3 各论中所述的安全数据建立的,适用于新药。各论中的PDE 未进行修约。为了实用的目的,本表中的PDE 已修约至1 位或2 位有效数字。PDE 小于10 的修约至最近的单位,保留1 位有效数字,适当时,大于10 的修约为1 位或2 位有效数字。本表中的修约原则可以用于其它给药途径的PDE 计算。
2 分类原则参见第4 部分定义。
表A.2.2:方法1 中元素杂质的允许浓度
本表中列出的值表示了药品、原料药和辅料中元素杂质的允许浓度,单位为mg/g。当选择使用方法1 时,可以使用这些浓度来评估日剂量不超过10g/天的药品中含有的元素杂质。本表中的数值是基于表A.2.1 的。
| 元素 | 分类 | 口服浓度 | 注射浓度 | 吸入浓度 |
|---|---|---|---|---|
| Cd 镉 | 1 | 0.5 | 0.2 | 0.2 |
| Pb 铅 | 1 | 0.5 | 0.5 | 0.5 |
| As 砷 | 1 | 1.5 | 1.5 | 0.2 |
| Hg 汞 | 1 | 3 | 0.3 | 0.1 |
| Co 钴 | 2A | 5 | 0.5 | 0.3 |
| V 钒 | 2A | 10 | 1 | 0.1 |
| Ni 镍 | 2A | 20 | 2 | 0.5 |
| Tl 铊 | 2B | 0.8 | 0.8 | 0.8 |
| Au 金 | 2B | 10 | 10 | 0.1 |
| Pd 钯 | 2B | 10 | 1 | 0.1 |
| Ir 铱 | 2B | 10 | 1 | 0.1 |
| Os 锇 | 2B | 10 | 1 | 0.1 |
| Rh 铑 | 2B | 10 | 1 | 0.1 |
| Ru 铷 | 2B | 10 | 1 | 0.1 |
| Se 硒 | 2B | 15 | 8 | 13 |
| Ag 银 | 2B | 15 | 1 | 0.7 |
| Pt 铂 | 2B | 10 | 1 | 0.1 |
| Li 锂 | 3 | 55 | 25 | 2.5 |
| Sb 锑 | 3 | 120 | 9 | 2 |
| Ba 钡 | 3 | 140 | 70 | 30 |
| Mo 钼 | 3 | 300 | 150 | 1 |
| Cu 铜 | 3 | 300 | 30 | 3 |
| Sn 锡 | 3 | 600 | 60 | 6 |
| Cr 铬 | 3 | 1100 | 110 | 0.3 |
附录3:单独安全评估(未译)
附录4:举例说明(未译)
Hits: 38
- 新原料药和制剂的稳定性试验
- 稳定性试验: 新原料药和制剂的光稳定性试验
- 稳定性试验:新剂型的要求
- 新原料药和制剂稳定性试验的括号法和矩阵法设计
- 稳定性数据的评价
- 分析方法验证:文本及方法学
- 新原料药中的杂质
- 新药制剂中的杂质
- 元素杂质指南
- 关于在ICH区域内评估并推荐采用药典相关要求的指导原则
- 关于ICH区域内药典附录的评价及建议-注射剂装量检查法
- 关于ICH区域内药典附录的评价及建议-不溶性微粒检查法
- 关于ICH区域内药典附录的评价及建议-非无菌产品的微生物检查:微生物计数法
- 关于ICH区域内药典附录的评价及建议-非无菌产品的微生物检查:控制菌检查法
- 关于ICH区域内药典附录的评价及建议-非无菌产品的微生物检查:原料药及其制剂的判定标准
- 关于ICH区域内药典附录的评价及建议 -崩解时限检查法
- 关于ICH区域内药典附录的评价及建议 -溶出度检查法
- 关于ICH区域内药典附录的评价及建议 —无菌检查法
- 关于ICH区域内药典附录的评价及建议 -片剂脆碎度检查法
- 关于ICH区域内药典附录的评价及建议 —聚丙烯酰胺凝胶电泳法通则
- 关于ICH区域内药典附录的评价及建议 —毛细管电泳法
- 关于ICH区域内药典附录的评价及建议 —筛分法
- 关于ICH区域内药典附录的评价及建议 —粉末的散装密度和振实密度测定法
- 来源于人或动物细胞系的生物技术产品的病毒安全性评价
- 生物技术产品的质量:rDNA衍生蛋白质产品生产细胞的表达构建体分析
- 质量标准 新原料药和制剂的检测以及可接受标准:化学物质
- 质量风险管理
- 药品研发
- 药物致癌试验的必要性
- 人用药物的遗传毒性试验和数据分析指导原则
- 毒代动力学指导原则:毒性研究中全身暴露的评价
- 动物慢性毒性试验的周期(啮齿类和非啮齿类)
- 药代动力学:重复给药的组织分布研究指导原则
- 药物生殖毒性和雄性生育力毒性的检测
- 生物技术药品的临床前安全性试验
- 人用药延迟心室复极化(QT间期延长)潜在作用的非临床评价指导原则
- 人用药物的安全性药理学研究
- 人用药品的免疫毒性研究
- 抗癌药物的非临床评价
- 药物的光安全性评价
- 对非危及生命的疾病的长期治疗药物进行临床安全性评估的人群暴露程度
- 临床安全性数据管理:快速报告的定义和标准
- 药物警戒计划
- 研发安全性更新报告
- 临床研究报告的结构与内容
- 新药注册所需的量-效关系资料
- 引入海外临床数据时要考虑的种族因素
- 老年人群的临床研究
- 临床试验的一般考虑
- 临床试验统计原则
- 儿童用药品的临床调查
- 新抗高血压药的临床评价原则
- 基因组生物标记物、药物基因组学、遗传药理学、基因组数据和样本编码分类的定义