
Contents
- 1 摘要
- 2 主要的
- 3 讨论
- 4 方法
- 4.1 试验设计和数据报告
- 4.2 下一代测序
- 4.3 生物信息学和突变发现
- 4.4 新表位优先排序和选择
- 4.5 RNA-LPX 的良好生产规范生产
- 4.6 用于免疫原性检测的血液采样
- 4.7 重叠肽
- 4.8 外周血单核细胞的 IVS
- 4.9 IFNγ ELISpot
- 4.10 肽-MHC 多聚体染色
- 4.11 细胞内细胞因子染色
- 4.12 肿瘤组织 TCR 谱分析
- 4.13 血液样本 TCR 测序和生物信息学分析
- 4.14 单细胞测序和读取处理
- 4.15 RevImMo 分析
- 4.16 单细胞基因表达分析
- 4.17 对多聚体阳性细胞进行单细胞 TCR 测序
- 4.18 免疫组织化学染色
- 4.19 差异基因表达分析、GSEA 和变异等位基因频率计算
- 4.20 Kaplan-Meier 生存分析用于评估无病生存概率
- 4.21 材料供应情况
- 4.22 报告摘要
- 5 数据可用性
- 6 参考
Individualized mRNA vaccines evoke durable T cell immunity in adjuvant TNBC
摘要
三阴性乳腺癌 (TNBC) 即使在早期也常发生转移性复发 1 。本研究评估了一种个体化新抗原 mRNA 疫苗在 14 例 TNBC 患者术后以及新辅助或辅助治疗后的疗效。在几乎所有患者的外周血中,均检测到了针对多种新抗原的高强度、疫苗诱导的、主要为新生 T 细胞应答,且这些应答持续数年。对个体患者的特征分析显示,这些 T 细胞中很大一部分分化为两个亚群:一个是具有“随时可发挥作用”的细胞毒性效应 T 细胞标志物的晚期分化表型,另一个是具有干细胞样记忆表型的 T 细胞。11 例患者在接种疫苗后长达 6 年内未出现复发。3 例患者出现复发:其中疫苗诱导的 T 细胞应答最弱的患者复发,但在随后的抗 PD-1 治疗后达到完全缓解;另一名患者的肿瘤主要组织相容性复合体(MHC)I 类分子表达较低,且在接种疫苗后,MHC I 类分子缺陷细胞仍持续生长;第三名患者为 BRCA 阳性,且原发肿瘤基因不同,出现复发。这些发现证明了个体化 RNA 疫苗在三阴性乳腺癌(TNBC)治疗中的可行性,证实了疫苗诱导的功能性新抗原特异性 T 细胞的持久性,并为可能的免疫逃逸机制提供了新的见解,这将指导未来的治疗策略。
其他人也在浏览类似内容



主要的
TNBC 即使在疾病早期也容易复发和转移,复发率在大约三年后达到高峰,之后迅速下降 1 。DNA 修复缺陷、内在基因组不稳定性以及免疫原性肿瘤微环境使 TNBC 成为使用可作为新抗原的体细胞癌突变进行个体化疫苗接种的有趣候选者 2 , 3 。
我们开发了一种个体化的新抗原 RNA 疫苗策略,该策略利用新一代测序(NGS)进行突变检测,并预测潜在的 T 细胞免疫诱导新抗原。目前正在对黑色素瘤、胰腺癌和其他实体瘤患者进行该策略的研究 4 , 5 , 6 , 7 (扩展数据图 1a )。为了构建该疫苗,我们将来自同一患者的多个癌症突变串联起来,并编码在两条免疫刺激性、非核苷修饰的尿苷 mRNA 链上。mRNA 的帽结构、5′和 3′非翻译区以及 poly(A)尾的分子设计增强了编码序列在人树突状细胞中的翻译 8 , 9 。我们将分泌信号和 MHC I 类转运结构域(MITD)标签融合到编码序列上,以增强编码抗原的人类白细胞抗原(HLA)I 类和 II 类分子呈递以及 CD8 + 和 CD4 + T 细胞的识别 10 。脂质体纳米颗粒(LPX)制剂用于静脉注射,靶向体内所有淋巴组织中的驻留树突状细胞 11,12 (图 1a )。RNA-LPX 疫苗技术中尿苷 mRNA 的应用,将抗原递送与 Toll 样受体(TLR)介导的 I 型干扰素驱动的抗病毒免疫反应的共刺激联系起来,从而显著扩增人体内抗原特异性 T 细胞,即使针对非突变自身抗原也有效 11,12,13 。
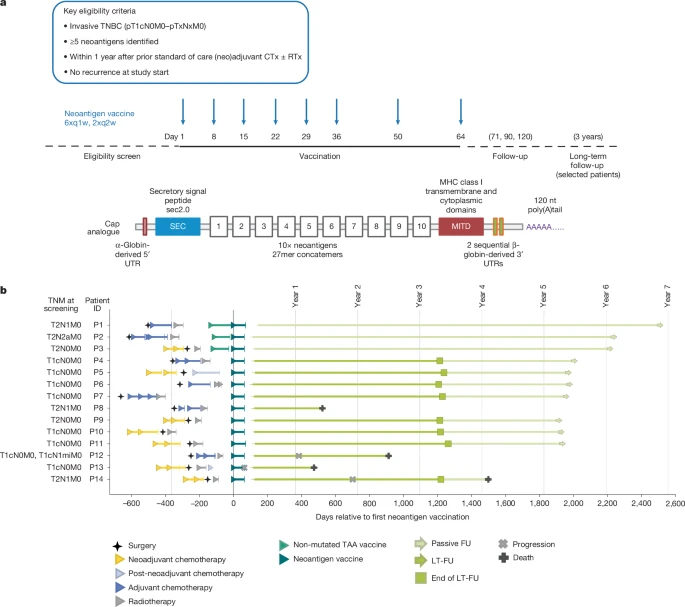
图 a ,顶部,临床试验设计。接受手术和新辅助及辅助化疗(CTx)(伴或不伴放疗(RTx))的早期三阴性乳腺癌(TNBC)患者,在接受标准治疗后一年内符合入组条件。中间,导入队列( n = 3,患者内剂量递增:14.4 µg、29 µg 和 50 µg,随后 5 × 50 µg)先于主要队列( n = 12)进行,主要队列患者接受 8 × 50 µg mRNA-LPX 疫苗治疗。导入队列患者接受预处理(根据患者肿瘤表达谱,从预先制备的包含八种非突变型肿瘤相关抗原(TAA)的仓库中组装的现成 mRNA-LPX 疫苗),作为过渡治疗,直至其个性化疫苗可用。底部所示为个体化新抗原 mRNA 疫苗,该疫苗由编码在两个多脂复合物 mRNA 分子上的 20 个新抗原靶点组成。对于单核苷酸变异(SNV),使用中心氨基酸发生改变的 27 肽段;对于移码突变引起的插入或缺失,则使用从发生改变的氨基酸到下一个终止密码子的序列。编码新抗原靶点串联体的开放阅读框两侧分别连接有分泌信号肽(SEC)和 MITD,以增强 HLA 呈递。mRNA 的 5′和 3′非翻译区(UTR)以及 poly(A)尾的设计均经过优化,以提高其稳定性和翻译效率。三名接受过桥接 TAA 疫苗预治疗的患者接受了被动随访 。b ,本试验入组患者的既往治疗、试验方案和疾病进程。在 15 名同意接受治疗的患者中,有 1 名患者在接种 3 次疫苗后因出现治疗期间出现的不良事件(低血压,3 级;恶心,2 级;寒战,1 级)而停止治疗,因此未在此处显示。 FU,随访;LT-FU,长期随访;q1w,每周一次;q2w,每周两次;TNM,肿瘤分期淋巴结和转移分级系统。
在探索性首次人体 TNBC-MERIT(突变组工程化 RNA 免疫疗法)伞状研究 14 ( ClinicalTrials.gov:NCT02316457 )的一个组中评估了新抗原疫苗。该研究以非对照的方式探索了各种 mRNA 癌症疫苗在早期 TNBC 患者中的可行性、安全性和诱导抗原特异性免疫反应的能力。符合条件的患者需在完成标准治疗后一年内入组。肿瘤组织用于突变检测和定制疫苗设计。患者接受 8 剂个性化新抗原疫苗(6 次每周一次,2 次每两周一次,最后一次在第 64 天),该疫苗由最多 20 个由 2 个 RNA-LPX 分子编码的癌症突变组成(图 1a )。15 名知情同意的患者中有 14 名接受了符合方案的治疗,并可进行疗效评估(图 1b 和扩展数据表 1 )。前三名患者初始接受 14.4 µg 剂量的 RNA,随后剂量逐渐增加至目标剂量 50 µg。这些患者同时接种了一种现成的基于 RNA-LPX 的疫苗,该疫苗编码非突变的、共享的肿瘤相关自身抗原(TAA),作为过渡疫苗,直至按需生产的个性化疫苗可用。随后的 11 名患者接种了 50 µg RNA 剂量的肿瘤新抗原疫苗,未接种过渡性 TAA 疫苗(扩展数据图 1 )。
14 例患者中有 5 例在初诊时淋巴结阳性(图 1b ),所有患者均接受了根治性手术(扩展数据表 1 )。7 例既往接受过新辅助化疗的患者中,3 例的病理完全缓解(pCR)状态可用——1 例达到 pCR(P14),2 例未达到 pCR(P3 和 P9)。从初诊到入组本试验的时间间隔为 0.56 至 1.53 年。按需生产在标准临床环境下被证实是可行的,所有符合治疗意向的患者均接种了其个体化疫苗。疫苗生产的平均周转时间(定义为从收到样本到疫苗发放的时间)为 69 天(范围:34-125 天)。值得注意的是,本研究的主要目标并非优化周转时间,而是利用该技术平台稳健地建立从手术到疫苗接种的端到端生产流程。
与研究药物相关的最常见治疗期间出现的不良事件(TEAE)是典型的反应原性症状,例如发热、头痛、寒战、恶心和疲乏(扩展数据表 2 ),这些症状通常在给药后的 1-3 天内出现,且大多为 1 级或 2 级。TEAE 具有短暂性,可用退热药控制,通常在一天内消退,这与之前基于该 RNA-LPX 平台的其他癌症疫苗的报道一致 6 , 11 , 15 。
我们采用两种互补的 IFNγ酶联免疫斑点(ELISpot)检测方法,评估了基线和第八剂(最后一剂)疫苗接种后 7-14 天采集的血液样本中针对新抗原的特异性 T 细胞反应。对于所有 14 名受试者,我们进行了体外 ELISpot 检测,该方法可将外周血单核细胞(PBMC)与覆盖每位受试者疫苗编码新抗原序列的重叠肽(OLP)孵育过夜,从而检测高强度、晚期分化的 T 细胞反应。其中 8 名患者在体外用负载覆盖疫苗编码新抗原 OLP 的自体抗原呈递细胞刺激 CD4 + 和 CD8 + T 细胞 1.5 周后,剩余的 PBMC 足以进行 ELISpot 检测。该方法可捕获低频、早期分化且增殖潜能高的 T 细胞,从而有助于检测 CD4 + T 细胞反应(其频率通常较低)。
所有 14 名患者均产生了针对 1 至 10 个疫苗靶点的疫苗诱导或增强的 T 细胞应答,这已通过至少一种 ELISpot 方法检测到(图 2a )。除 1 名患者外,其余患者的应答均针对多种疫苗抗原。9 名患者对至少 5 种新抗原产生了应答(例如,图 2a 中患者 P12 对 8 种疫苗新抗原产生了新生 T 细胞应答)。12 名患者(86%)的应答可通过体外 ELISpot 检测。这 12 名患者中有 8 名对单一新抗原的应答达到每 10⁶个 PBMC 中 2000-4000 个 IFNγ⁺ 斑点(图 2a、b 和扩展数据图 2a )。在接种疫苗前,针对 14 名患者使用的 251 个突变中的 41 个突变的 T 细胞无法检测到,但在接种疫苗后可以检测到。基线时存在针对两种新抗原的 T 细胞,接种疫苗后进一步扩增(图 2c ,左上和补充数据 1 )。四名个体表现出针对已知癌症驱动基因( CREBBP 、 DAXX 、 IGF1R 或 TP53 )的新生反应(补充数据 1 ,第二个标签页)。

a ,通过体外 IFNγ ELISpot 检测患者 (P1-P14) 外周血单核细胞 (PBMC) 中新抗原疫苗诱导的 T 细胞反应数量( n = 14)和静脉注射疫苗后 (IVS) 检测的数量( n = 8 ) 。柱状图上方显示了免疫原性突变的总数。b,左图:所有参与者通过体外 IFNγ ELISpot 检测的针对单个新抗原的 T 细胞反应强度。右图:P11 的 ELISpot 数据。TNTC:数量过多,无法计数。c,IFNγ ELISpot T 细胞反应。上图 :体外检测(左图;14 名患者共检测到 251 个疫苗编码突变)和静脉注射疫苗后检测(右图;由于样本量有限,仅检测了 8 名患者共检测到的 115 个疫苗编码突变中的 85 个)。下图:比较体外和静脉注射疫苗后 SNV(实线柱)和插入缺失(虚线柱)反应占可评估靶标的百分比。 d ,通过 IFNγ ELISpot 检测个体(左)和队列(中,显示阳性反应)水平的 IVS 后 T 细胞反应类型。右图为 P7 的 ELISpot 数据。a – d 中的数据为末次接种疫苗后 7-14 天检测到的 T 细胞反应 。e ,每位患者疫苗诱导的多新抗原反应强度随时间的变化,显示体外 IFNγ + ELISpot 检测中每种免疫原性疫苗新抗原的斑点总数(范围:2-8)。对于 P14,仅能检测两种疫苗诱导反应的新抗原中的一种 。f ,通过 HLA 多聚体染色对 P1 的十种新抗原特异性 T 细胞反应中的两种进行表征。左图为针对两种新抗原特异性疫苗抗原的 CD8 + T 细胞。正确,用代表相应新抗原的肽刺激 PBMC 中多聚体阳性新抗原特异性 CD8 + T 细胞的细胞内细胞因子染色。 g 、 h :通过多聚体染色法测定疫苗诱导的新抗原特异性 CD8 + T 细胞的累积频率( g )和表型( h )(10 种新抗原,6 例患者)。曲线代表 P1、P10、P11 和 P12 对两种新抗原的多聚体数据之和;P6 和 P7 仅对一种抗原有反应。在 e – h 中,虚线表示疫苗接种。
对 8 名患者用于疫苗接种的 85 个突变进行 IVS 后 ELISpot 分析,鉴定出针对其中 44 个(51.8%)突变的特异性 T 细胞。超过一半( n = 26)是新诱导的(图 2c ,右上)。在 IVS 后 ELISpot 检测中,约三分之二(44 个中的 28 个,64%)具有免疫原性的新表位被 CD4 + T 细胞识别,20%(44 个中的 9 个)被 CD8 + 细胞毒性淋巴细胞 (CTL) 识别(图 2d ),16%(44 个中的 7 个)同时被 CD4 + 和 CD8 + T 细胞识别,如患者 P8 所示,他具有预先存在的疫苗增强的 CD4 + T 细胞反应和针对新抗原 KIAA0564(R664L) 的新生诱导的 CD8 + T 细胞反应(扩展数据图 2b )。
疫苗诱导的 T 细胞的突变特异性已在一名患者 (P1) 中得到证实(扩展数据图 2c ),并且此前已使用这种新抗原 mRNA 疫苗技术得到证明 4。 单核苷酸变异 (SNV) 以及插入/缺失突变 (indel) 可诱导 T 细胞;indel 能够提供多个表位,例如患者 P3,其针对 KDM5A 移码缺失衍生的不同 MHC I 类等位基因上呈现的独立表位表现出 T 细胞反应(扩展数据图 2d 和补充表 1 )。
共有 9 名患者(共携带 27 个免疫原性突变)在长期随访期间提供了血样,因此可通过体外 ELISpot 法评估疫苗诱导免疫反应的持久性。患者 P5、P8、P6、P10、P11 和 P12 的疫苗诱导多新抗原免疫反应(各疫苗新抗原免疫反应强度的总和)经历了快速扩增和短暂下降,随后在 1 至 3.5 年内维持在高水平(图 2e 和扩展数据图 3 )。患者 P7 和 P9 的新抗原特异性 T 细胞扩增和持久性较弱,而患者 P14 的反应则非常弱。在个体新抗原特异性 T 细胞反应层面,除一名个体(P7)外,所有个体中至少有一种疫苗诱导的 T 细胞在没有疫苗加强针的情况下至少 12 个月仍可检测到(图 2e 和扩展数据图 3 )。
对于 6 例体外反应可检测到的患者,我们能够针对其中两种疫苗诱导的 T 细胞特异性(P6 和 P7 仅一种)生成功能性肽-MHC I 类复合物,并通过多聚体染色证实了 CD8 + T 细胞反应的动力学。如图 2f 所示(P1 在单个新抗原水平上的情况;扩展数据图 7a 中的设门策略),以及图 2g 所示(所有 6 例患者的累积多新抗原反应),新诱导的突变特异性 CD8 + T 细胞在疫苗接种期间迅速扩增,在接种 3 剂疫苗后(约第 3 周)即可检测到,并在最后一次接种疫苗后,其在所有循环 CD8 + T 细胞中的频率中位数达到 6.58%(范围:0.5%–17.5%)。在最后一次疫苗接种后 1 至 6 年,血液中仍能检测到几种新抗原特异性 CD8 + T 细胞,其比例为个位数百分比(图 2g )。例如,在疫苗接种方案结束时,P1 患者循环 CD8 + T 细胞中有 10.3%识别 HEATR2(R47Q),这是该患者针对其产生新生免疫应答的 10 种突变之一。两年后,尽管未进行疫苗加强免疫,这些细胞(仅代表 P1 疫苗诱导的多新抗原 T 细胞应答的一小部分)仍占循环 CD8 + T 细胞的 3%以上(图 2f )。P6 患者在接种疫苗后,针对 MRPS5(S194I)的新生诱导 T 细胞比例为 17.5%,并在 3 年内下降至 1.5%(图 2g )。 疫苗诱导的 CD8 + T 细胞在最初几个月内大多具有效应记忆表型,并在 3 至 6 年内发展为更晚期分化的 CD45 再表达表型(图 2h ),但 P12 除外,在 4 次疫苗接种后,几乎所有新抗原特异性 T 细胞都具有晚期分化表型(图 2h )。
由于 P1 患者长期缓解(接种疫苗后超过六年未复发),且在此期间收集了外周血单核细胞(PBMC),因此该患者成为详细分析疫苗诱导的 T 细胞反应的理想病例。P1 患者针对 10 种疫苗靶向突变产生了 T 细胞反应,包括针对 8 种新抗原的强烈的体外 IFNγ分泌反应,每百万 PBMC 中约有 9000 个斑点形成单位(图 2b ,左图和扩展数据图 2a )。在体外免疫抑制(IVS)后,还检测到针对另外两种新抗原的较低强度的反应,这些反应由 CD4 + T 细胞介导(图 2a 和补充表 1 )。
为了解析 P1 的 T 细胞库,我们采用了反向免疫监测(RevImMo)技术,该技术结合了整体和单细胞 T 细胞受体(TCR)测序,以识别疫苗接种后扩增的新抗原特异性克隆型。我们克隆了在疫苗接种后单细胞 TCR 测序数据中富集的 26 个克隆,并使用增强型 Jurkat-NFAT 报告基因检测方法 6 (图 3a )进行了检测。在 26 个富集程度最高的具有配对 TCR 序列的 CD8 + T 细胞克隆中,有 16 个能够识别由主要组织相容性复合体 IB 类(HLA-B)等位基因呈递的三种 ELISpot 阳性疫苗靶点——HEATR2(R47Q)、PPP1R15B(S278T)和 PTCD3(I448T)(图 3b 和扩展数据图 2a 和 4a )。其中 11 个 TCR 是新产生的;其余 5 个是先前存在的。

本文介绍了一种基于 RevImMo 的方法,用于鉴定和功能表征疫苗扩增的新抗原特异性 T 细胞克隆。该方法首先对接种疫苗前后的 CD8 + 或 CD4 + T 细胞进行批量 TCR 测序。然后,计算每种 TCRβ 链 (TRB) CDR3 克隆型的治疗后富集度。利用治疗后 CD8 + 或 CD4 + T 细胞的单细胞 TCR 测序数据,检索相应的配对 TCRα 链 (TRA) 和 TRB。富集分析仅考虑具有配对链数据的克隆型,并选择最多 30 个富集度最高的克隆型进行 TCR 克隆和体外特异性测试。将编码 TCR 链的 RNA 转染到 Jurkat 效应细胞中,并与转染了个体 HLA I 类或 II 类分子并负载了代表其疫苗新抗原靶点的肽段的抗原呈递细胞进行共培养。 TCR 反应性通过评估 NFAT 诱导的荧光素酶报告基因活性来确定。UMAP,均匀流形近似和投影。使用 BioRender 创建。Cortini, A. (2025) https://BioRender.com/6smlkd5。b、c,P1 的 RevImMo 分析和 TCR 动力学。左上,疫苗接种前后血液中 CD8 + 和 CD4 + T 细胞克隆的总体 TCR 谱数据。RevImMo 选择的克隆已突出显示;疫苗新抗原特异性 TCR 以实心圆表示,特异性未确定的 TCR 以空心圆表示。追踪纵向血液样本中 CD8 + ( b ,右上)和 CD4 + ( c ,右)总体 TRB 谱数据中的新抗原特异性克隆。b , 底部,所有 TCR 的频率及其按特异性的分离。 d ,对 P1、P12 和 P13 纵向血液样本中的新抗原特异性 CD8 + T 细胞进行无监督聚类分析(左上),并展示每个新抗原特异性细胞簇中差异表达最显著的 10 个基因(右上)。 底部 ,纵向血液样本中新抗原特异性细胞亚群的比例。e,通过多聚体染色鉴定患者 P1 新抗原特异性 CD8 + T 细胞群中 TCF-1 + IL-7Rα + 的频率。虚线表示疫苗接种情况。
通过 CDR3 序列追踪发现,在 P1 受试者接种疫苗后 6 年内,新抗原特异性 CD8 + T 细胞持续存在于 CD8 + TCR 库中约 1.9%的水平(图 3b ),这与 PPP1R15B(S278T)和 HEATR2(R47Q)的多聚体分析结果一致(图 2f ,右)。利用细胞内细胞因子染色,我们发现这两种持久存在的 T 细胞群在患者最后一次接种疫苗后长达 3.5 年的时间里持续产生 TNF 和 IFNγ(图 2f ,右和扩展数据图 7b、c )。此外,在接种疫苗后分别长达 4.5 年和 6 年的时间里,还检测到了两种识别不同疫苗靶点的低丰度 CD4 + TCR(图 3c )。
我们将 TCR 库水平分析扩展至患者 P12 和 P13,利用纵向单细胞 RNA 测序(RNA-seq)和 TCR 测序数据集(扩展数据图 5a、b )中鉴定的 TCR 序列追踪新抗原特异性 CD8 + T 细胞。对总 CD8 + T 细胞群进行无监督聚类分析,揭示了九个转录亚群,分别代表初始 T 细胞(TN)、中央记忆 T 细胞( TCM )、三个效应记忆 T 细胞亚群( TEM1 至 TEM3 )、两个终末分化效应 T 细胞亚群( TEMRA1 和 TEMRA2 )、黏膜相关不变 T 细胞(MAIT)以及一个增殖性 T 细胞簇(扩展数据图 5a )。在这些亚群中,在最后一次疫苗接种后 1 至 2 周,新抗原特异性 CD8 + T 细胞主要表现出重新表达 CD45RA (T EMRA ) 的终末效应记忆表型,并且在整个随访期间一直作为主要亚群存在(扩展数据图 5b )。
我们在新抗原特异性 CD8 + T 细胞库中鉴定了五个转录不同的簇:一个 T EMRA 簇,以 GNLY 、 ZNF683 和其他与细胞毒性和组织驻留相关的基因为标志;一个 T EM 簇,表达 KLRG1 、 LAIR2 和 CCL5 ,与记忆样特征一致;一个活化的效应记忆 T 细胞 (T eff ) 群,产生细胞因子 ( CCL4 、 TNF 和 IFNG ),表明具有活跃的效应功能;一个增殖性 T (T prolif ) 细胞群,以细胞周期基因 ( UBE2C 和 KIF2C ) 为特征;以及一个干细胞样记忆表型 (T SCM ) 亚群,共表达 CCR7 、 TCF7 、 LEF1 、 IL7R 和其他干细胞样记忆和幼稚谱系的标志物(图 3d ,顶部)。治疗结束时(第 70-85 天)的早期反应主要由 T EM 和 T eff 细胞构成(图 3d ,底部和扩展数据图 5d、e )。随着时间的推移,T eff 细胞逐渐被 T EMRA 细胞所取代,后者与 T EM 细胞一起成为长期随访期间的主要表型。值得注意的是,干细胞样记忆 T SCM 细胞群也出现并持续存在,这支持了持久且表型多样化的记忆 T 细胞库的存在(图 3d ,底部和扩展数据图 5d、e )。在 P1 组中,我们利用流式细胞术证实,在接种疫苗后 6 年以上,疫苗诱导的干细胞样新抗原特异性 T 细胞持续存在,这些细胞共表达 TCF-1 (也称为 TCF7 )和 IL7RA (也称为 IL7R )(图 3e 和扩展数据图 5d、e)。 5c ),强调了具有再生潜能的记忆性 T 细胞库的持久性。
截至 2025 年 2 月,14 例患者中,10 例在末次疫苗接种后中位随访 5 年(中位数为 62 个月,范围 15-80 个月)内未出现复发(图 1b 和扩展数据图 6 );此外,患者 P8 在末次疫苗接种后 15 个月因不明原因死亡前也未出现复发。患者 P12、P13 和 P14 出现疾病复发。在所有接受治疗的患者中,P14 的疫苗诱导免疫反应最弱。体外 ELISpot 数据显示,末次疫苗接种后不久,针对两种疫苗新抗原的 T 细胞数量级比大多数其他患者低一个数量级,但在后续时间点未观察到此现象;此外,静脉注射后捕获了针对另外三种疫苗新抗原的疫苗扩增 CD4 + T 细胞(图 2b 左和扩展数据图 3 )。在接种最后一剂疫苗 20 个月后,P14 的右侧乳房和腋窝出现局部复发。新病灶浸润了 T 细胞,肿瘤细胞表现出强烈的 MHC I 类分子表达(扩展数据图 8a )。该患者接受了抗 PD-1 抗体和序贯化疗,并获得了完全缓解,缓解期持续了 15 个月。之后出现全身性复发,患者于 3 个月后死亡。在接种最后一剂疫苗一周后,该患者体内检测到的三个疫苗诱导的 CD4 + 免疫应答持续到第 867 天,此时正值复发后且处于抗 PD-1 治疗期间(扩展数据图 2e )。另外两名肿瘤复发的患者(P12 和 P13)针对多个疫苗靶点产生了高强度的疫苗诱导免疫应答(图 8a)。 2b ,左)。
患者 P12 患有 BRCA1 基因突变阳性的双侧三阴性乳腺癌 (TNBC),接受了双侧肿瘤切除术,右乳肿瘤分期为 T1cN0,左乳肿瘤分期为 T1cN1,术后接受了辅助化疗和个体化新抗原疫苗接种。对右乳肿瘤进行了基因测序,以指导疫苗设计。体外 ELISpot 实验证实(图 2b ,左图和补充数据 1 ),P12 针对八个靶点产生了强效的新抗原特异性 T 细胞反应。通过 RevImMo(图 4a 和扩展数据图 4b )以及对多聚体富集细胞群进行单细胞 TCR 测序(扩展数据图 8b ),我们鉴定出 107 个新抗原特异性 TCR,涵盖 4 个不同的抗原-HLA 对:GPR39(I173M)-HLA-A03:01、IGF1R(G108D)-HLA-B15:01 和 PEX6(L409F)-HLA-B15:01 和 HLA-C03:03。这些 TCR 在疫苗接种前均不存在,这与强烈的、多克隆的新生免疫应答相符。表型分析显示,以 T EM 细胞应答为主,并随时间推移逐渐分化为记忆干细胞样 T SCM 细胞(图 3d ,底部和扩展数据图 5b )。在接种疫苗两到三个周期后,首次检测到靶向免疫优势新抗原之一的 T EMRA 细胞,且这些细胞长期存在(图 2h )。尽管克隆广度得以维持,但在最后一次接种疫苗后的两个月内,TCR 频率显著降低(图 4a ,扩展数据图 8b )。十个月后,患者出现复发,最初表现为 BRCA1 阳性的胸腔淋巴结转移,随后扩散至肺部和脑部。该患者在接种疫苗后 17 个月因疾病进展而死亡。 值得注意的是,转移灶保留了强烈的 MHC 表达(扩展数据图 8c ),但测序的右侧乳腺肿瘤中的体细胞 SNV 均未出现(扩展数据图 8d )。相反,有 61 个 SNV 与左侧乳腺肿瘤重叠,表明复发可能起源于这个独立演化的肿瘤(扩展数据图 4b )。与此起源相符的是,在复发转移灶中未检测到新抗原特异性 TCR(扩展数据图 8e )。

a 、 c ,P12 ( a ) 和 P13 ( c ) 中新抗原特异性 T 细胞克隆的特征分析。通过比较接种疫苗前后的血液样本,利用 RevImMo 分析评估疫苗诱导的新抗原特异性 T 细胞克隆扩增。左上图为 TCR 谱分析散点图;图中显示了具有新抗原特异性 TCR 的克隆(实心圆)和具有未解析特异性 TCR 的克隆(空心圆)。柱状图追踪了纵向血液样本 CD8 + TRB 谱分析数据中的新抗原特异性克隆,显示了所有 TCR 的累积频率(右上)以及按特异性划分的频率(下) 。b ,比较从患者 P12 获得的不同肿瘤组织样本中检测到的 SNV。d , 上图显示了治疗前和复发肿瘤组织之间的差异表达基因。浅绿色表示 | log2 (倍数变化)| 值大于 0.05 的基因。 > 1 和 q 值 < 0.01(Wald 检验,双侧,经 Benjamini-Hochberg 程序校正);深绿色,京都基因与基因组百科全书 (KEGG)“抗原加工和呈递”通路基因集富集分析 (GSEA) 的前沿基因(底部); NES ,归一化富集评分。e,P13 患者接种疫苗后复发肿瘤组织和接种疫苗前肿瘤(单染)的福尔马林固定石蜡包埋 (FFPE) 切片的染色。插图显示结肠组织染色作为阳性对照。比例尺,50 μm。f ,P13 患者血液和肿瘤中 CD8 + T 细胞新抗原特异性 TCR 的累积频率。疫苗接种前血液与原发肿瘤(左图)以及疫苗接种后血液与复发肿瘤(右图)之间存在共享克隆型。TRB CDR3 序列用于批量分析数据的追踪。 条形图上方的数字:检测到的新抗原特异性 TCR 数量与已鉴定的总数之比。
患者 P13 在完成疫苗接种后不久,右侧乳腺出现局部复发,并于 13 个月后死亡。RevImMo 分析显示,存在 10 个 CD8 + T 细胞克隆,这些克隆特异性针对体外 ELISpot 鉴定的 6 种疫苗新抗原中的 4 种——ADD2(Y304S)、SLC25A18(C229Y)、SSH1(R96C)——以及一个 TP53 插入/缺失突变(图 2b 和 4c ,扩展数据图 8f ,补充表 2 和补充数据 1 )。这些疫苗诱导的 T 细胞在表型上与患者 P1 的 T 细胞相似,但 TEM 细胞亚群的富集程度更高(图 3d 和扩展数据图 5b )。复发肿瘤中大多数疫苗新抗原的变异等位基因频率增加,包括那些驱动强 T 细胞反应的抗原(扩展数据图 8f、g )。然而,转录组分析显示 , 与原发肿瘤相比,参与抗原呈递的关键基因(包括 MHC I 类分子和 β2 微球蛋白) 表达下调(图 4d 和补充数据 2、3 )。免疫组化染色显示,原发肿瘤 MHC I 类分子表达低且 T 细胞浸润极少,而复发肿瘤 MHC I 类分子几乎完全缺失,免疫细胞浸润进一步减少(图 4e )。对血液和肿瘤组织进行 TCR 谱分析表明,接种疫苗前,血液中存在三种低频率的新抗原特异性 TCR,但在原发肿瘤中未检测到。接种疫苗后,鉴定出十种新抗原特异性 TCR,其中六种克隆型存在于复发肿瘤中(图 4f )。 尽管有功能性疫苗诱导的 T 细胞浸润,但复发性肿瘤可能通过下调抗原呈递途径来逃避免疫压力。
讨论
我们的研究表明,在接受佐剂或新佐剂治疗的三阴性乳腺癌(TNBC)患者中,个体化尿苷 mRNA 新抗原疫苗具有临床可行性、安全性和强大的免疫原性。该疫苗耐受性良好,几乎所有患者均能针对其多种新抗原产生强烈的特异性 T 细胞应答。在个体患者中,外周血中多表位免疫应答的 T 细胞数量达到两位数,与过继性 T 细胞疗法通常观察到的水平相当。
我们观察到多个病例,相同的突变序列产生了可被 CD4 + 和 CD8 + T 细胞同时识别的新抗原表位,或者新抗原表位通过不同的 HLA I 类限制性元件呈递给不同的 CD8 + T 细胞克隆。接种疫苗后数年,外周血中仍可检测到处于功能状态的新抗原特异性 T 细胞,这凸显了持久免疫的潜力。值得注意的是,这些 T 细胞不仅持续存在,而且还经历了进一步的表型演变。其中相当一部分 T 细胞成熟为表达 GLNY 和 ZNF683 的晚期分化细胞毒性效应细胞,这些细胞是与良好临床结果相关的“随时可发挥作用”细胞的标志 16,17,18 。
同时,一部分早期分化的 T 细胞保留了高水平的干细胞样记忆标志物,如 IL-7Rα 和 TCF7(由 TCF-1 编码),TCF7 是一种转录因子,是响应病毒或肿瘤抗原而产生的干细胞样 CD8 + T 细胞自我更新所必需的,也是维持对检查点阻断免疫疗法的增强反应所必需的 19 。
我们观察到,疫苗设计中筛选出的癌症突变能够频繁诱导 CD4 + T 细胞的产生。然而,疫苗诱导的 CD4 + T 细胞反应的克隆频率明显低于 CD8 + T 细胞,并且由于外周血单核细胞数量有限以及技术上的限制,我们将研究重点放在了 CD8 + T 细胞上。
患者 P12、P13 和 P14 的研究结果提示了治疗失败和新抗原疫苗逃逸的不同可能机制。其中一种机制可能是最初缺乏有效的疫苗诱导免疫反应,正如患者 P14 的情况,他在这方面尤为突出。尽管该患者在接受抗 PD-1 治疗后获得了完全缓解(这在三阴性乳腺癌患者中极为罕见),但随后死于全身性复发。P12 和 P13 尽管产生了强烈的多新抗原免疫反应,但仍出现了肿瘤复发。
患者 P13 在初始肿瘤中 MHC 表达极低,复发时 MHC I 类分子丢失,这很可能是由 β2M 下调驱动的。HLA I 类分子呈递的完全丧失仍然是一种有效的逃逸机制,并且在几种强效的基于 T 细胞的疗法中均有观察到 4 , 20 , 21 , 22 , 23 ,这凸显了联合疗法的必要性,例如使用肿瘤靶向抗体来抑制 HLA 缺陷型癌细胞的过度生长,或使用免疫调节方法来恢复 β2M 缺陷型肿瘤中的新抗原识别 24 , 25 。
第三例复发患者 P12 患有 BRCA 突变的三阴性乳腺癌(TNBC),这种乳腺癌常伴有独立演化的无关的同步或异时性原发肿瘤 26,27,28 。P12 患者的双侧乳腺肿瘤(仅其中一个用于疫苗设计)在诊断时被发现是克隆独立的。对于存在抗原性不同的原发肿瘤风险的遗传易感人群,对多个病灶进行测序可能是明智之举。
该研究的局限性包括参与者人数较少,尤其是在进行更深入的免疫学分析的队列中,以及缺乏对照组来进一步研究可能的免疫逃逸。
TNBC 研究扩展了我们之前在其他肿瘤类型中的研究结果,表明这种个性化疫苗策略具有广泛的应用前景。在黑色素瘤中,我们证实了转移事件累积发生率的显著降低,表明 mRNA 新抗原疫苗适用于高突变负荷的肿瘤 4 。在突变数量少且免疫抑制环境高度严密的胰腺癌中,该疫苗在 50% 的患者中诱导了免疫应答,且与临床疗效呈正相关 5 。在突变负荷低至中等的 TNBC 中,我们证实所有患者均产生了新抗原特异性 T 细胞应答,其中超过 85% 的应答强度较高。TNBC 的预后较差,尤其是在新辅助化疗后未达到病理完全缓解的患者中 29,30,31 。新抗原疫苗在大多数患者中表现出的强免疫原性、诱导的 T 细胞应答的持久性以及早期临床疗效,均提示有必要在该患者群体中进行进一步的临床试验。
方法
试验设计和数据报告
这项开放标签、首次人体试验、I 期、三组对照的伞式试验( ClinicalTrials.gov:NCT02316457 )的主要目标是分别评估两种不同的基于 mRNA-LPX 的疫苗的可行性、安全性和耐受性:一种是由非突变 TAA 组成的现成疫苗,另一种是按需生产的个体化新抗原疫苗。疫苗诱导的抗原特异性免疫反应是次要终点。本文报告了该试验中个体化新抗原疫苗组的相关结果。该试验在德国和瑞典进行,符合《赫尔辛基宣言》和良好临床实践指南,并获得了独立伦理委员会(德国莱茵兰-普法尔茨州医师协会伦理委员会,美因茨;瑞典区域伦理审查委员会,乌普萨拉)和主管监管机构(德国保罗·埃尔利希研究所,朗根;瑞典药品管理局,乌普萨拉)的批准。所有患者均签署了书面知情同意书。
入组标准为:组织学确诊的浸润性腺癌三阴性乳腺癌(TNBC,pT1cN0M0–[any]T[any]NM0);既往接受过标准治疗(即原发肿瘤新辅助化疗后行手术或手术联合辅助化疗);允许既往接受过放疗;年龄至少 18 岁;造血、肝肾功能良好;肿瘤表达至少 5 种新抗原。患者需在完成标准治疗(例如手术和/或化疗和/或放疗)后一年内符合入组条件(具体治疗方案根据当地政策而定)。主要排除标准为:试验治疗开始前乳腺癌复发,以及存在临床相关的自身免疫性疾病或活动性病毒感染。个体化新抗原疫苗由两个单链 RNA 分子组成,每个分子编码至多 10 个新抗原靶点,这些靶点的选择基于每位患者肿瘤的体细胞突变分析。 RNA 被脂质体化制备成 RNA-LPX,用于静脉给药,具体方法见文献 11。 对于接受新辅助化疗的患者,使用诊断性穿刺活检的 FFPE 肿瘤样本进行肿瘤抗原表达分析。在特殊情况下(例如,样本质量差),可以使用手术切除的 FFPE 肿瘤组织进行抗原表达分析。使用 agCapture 3.4.2.6(ArisGlobal)进行电子数据采集。
2021 年春季,主要研究阶段(直至治疗结束及 2020 年 56 天随访期)评估的主要和次要终点的临床试验报告已提交给卫生部门。此后,接受桥接 TAA 疫苗预处理的三名患者接受了被动随访;其中一名患者同意通过一项研究项目提供血液样本。接下来的 11 名患者(仅接受新抗原疫苗治疗)参与了为期 3 年的主动长期随访,直至 2023 年。长期随访期间产生的数据已汇总在临床试验报告的附录中,并于 2024 年春季提交给卫生部门。此后,这些患者接受了被动随访。
下一代测序
使用改良版的 QIAamp DNA FFPE 组织试剂盒(Qiagen),从 3 份 10 μm 厚的 FFPE 肿瘤组织样本中提取肿瘤 DNA,每个样本重复两次。使用 AmpTec 公司的 ExpressArt Clear FFPE RNAready 试剂盒提取 RNA,每个样本重复两次。使用 DNeasy 血液和组织试剂盒(Qiagen)从 PBMC 细胞中提取 DNA。提取的核酸用于构建各种 NGS 文库。使用 NEBNext RNA 第一链合成模块和 NEBNext 超定向 RNA 第二链合成模块进行 cDNA 合成,并使用改良版的 SureSelect XT V6 Human All Exon 试剂盒(Agilent),以 100 ng 总 RNA 为模板,从 FFPE 肿瘤组织 RNA 构建靶向 RNA 测序文库,每个样本重复两次。使用改良版的 SureSelect XT V6 Human All Exon(Agilent)试剂盒,以 100 ng FFPE 肿瘤 DNA 和匹配的 PBMC DNA 为模板,构建 DNA 全外显子组文库,每个样本重复两次。肿瘤和匹配的 PBMC 的全外显子组测序 NGS 文库的制备方法如下:将 100 ng 基因组 DNA 在 15 μl 的反应体系中,使用 microTUBE-15 AFA Beads Screw-Cap(Covaris)管将片段化至平均长度约为 150 bp。NGS 文库稀释至 3 nM,并使用 Illumina HiSeq 3000/4000 PE Cluster Kit 以 10 pM 的浓度进行簇集。FFPE 和 PBMC DNA 的混合外显子组文库在三个泳道中进行四重测序,而 RNA 文库的重复样本则在一个泳道中进行双重测序。所有文库均使用两套 HiSeq 3000/4000 SBS 50 循环试剂盒(Illumina)在 Illumina HiSeq 4000 平台上进行 50 nt 双端测序。对于 P12,疫苗接种后的样本则使用 Illumina NovaSeq 6000 平台,采用 Illumina NovaSeq 6000 S2 流动槽和 Illumina NovaSeq 6000 S2 试剂盒 v.1.5(100 循环)进行 100 nt 双端测序。
生物信息学和突变发现
所有基因组学相关的数据分析步骤均由专有的生物信息学流程(BioNTech)协调完成,该流程采用 Python 编程语言编写,具体内容简述如下。对于每个 DNA 文库的重复样本,至少需要 180 × 10⁶ 条 50 nt 双端测序序列,覆盖≥70%的目标碱基,且测序深度≥100×。对于 RNA 文库,则至少需要 75 × 10⁶ 条 50 nt 双端测序序列。
为了进行突变检测,使用 bwa (v.0.7.10) 32 将 DNA reads 比对到参考基因组 hg19。使用 SAMtools (v.0.1.19) 33 将得到的比对文件转换为 BAM 格式。使用内部突变检测器 4 检测体细胞 SNV,并使用 Strelka 34 检测短插入和缺失,方法是将肿瘤 DNA 的比对 reads 与匹配的 PBMC DNA 的比对 reads 进行比较。
将已鉴定的体细胞变异的基因组坐标与 UCSC 已知基因转录本坐标进行比较,以将变异与基因、转录本和潜在的氨基酸序列改变关联起来。同义突变和无义突变被过滤掉。对于已验证的非同义癌症突变(被选为潜在的新抗原疫苗靶点),基于突变转录本序列确定突变肽序列(MPS),用于设计 MPS 串联疫苗。对于单核苷酸变异(SNV),这些是 27 个氨基酸的肽段区域,其中改变的氨基酸位于中心。对于导致移码的插入和缺失,MPS 包含从改变的氨基酸到下一个终止密码子的序列(最多 50 个氨基酸)。使用 SAMtools 基于匹配的 PBMC DNA 鉴定突变肽段区域的种系变异。首先根据 RNA-seq 读取结果确定蛋白质改变种系变异的相位,如果与体细胞突变同相,则将其纳入患者特异性新抗原疫苗靶点。
对于 RNA-seq,使用 sailfish (v.0.7.6) 33 将 RNA reads 比对到 hg19 转录组,以估计转录本的表达量。过滤掉未表达的转录本。
此外,使用 STAR (v.2.4.2a) 35 将 RNA 读段比对到 hg19 参考基因组,以确定体细胞突变与种系突变的相位,以及确定突变转录本相对于未携带体细胞突变的转录本的相对表达量。
使用 NGSCheckMate (v.1.0) 36 来确认在同一患者 ID 下分析的所有 DNA 和 RNA 文库的来源是否相同。
新表位优先排序和选择
使用 IEDB T 细胞预测工具(v.2.13)37 预测 HLA 结合亲和力。对于 HLA I 类,使用 IEDB 推荐的方法预测 MPS 中所有包含变异的 8-11 个碱基对 HLA-A/B/C 的亲和力。对于 HLA II 类,使用 consensus3 方法预测 MPS 中所有包含变异的 15 个碱基对 HLA-DRB 的亲和力。在所有单个变异的预测结果中,选择最佳的共识评分与相应的 MPS 相关联。
使用相同的内部生物信息学流程,并采用下述排序和筛选方法,对所有已识别的 MPS 进行优先级排序,最多筛选出 46 个 MPS。首先,仅考虑 RNA 中变异等位基因频率>0 的体细胞突变。然后,从这些突变中:(1)根据 HLA I 类结合亲和力,选择最多 5 个插入或缺失突变的 MPS;(2)根据 HLA II 类结合亲和力和转录表达≥10 RPKM,选择最多 20 个 SNV 突变的 MPS;(3)根据 HLA I 类结合亲和力和转录表达≥1 RPKM,选择最多 20 个 SNV 突变的 MPS;(4)根据转录表达,进一步选择 SNV 突变的 MPS,直至总共达到 46 个 MPS;如果最终选择的 MPS 少于 46 个,则根据以下条件添加 RNA 中变异等位基因频率为零的体细胞突变:(6)HLA I 类评分;(7)转录表达。
我们使用 R 语言实现的算法,根据 HLA I 和 HLA II 结合预测、转录本表达、变异等位基因频率以及其他标准,从优先排序的 MPS 列表中筛选出至多 20 个新抗原疫苗靶点。每个患者的筛选结果均由审查委员会进行人工审核。
RNA-LPX 的良好生产规范生产
2019 年期间,所有患者均进行了生产运行。由于新抗原 RNA-LPX 的个性化按需生产流程是作为该临床试验的一部分启动的,并在患者入组期间不断优化,因此没有预先设定周转时间目标。
简而言之,两个分别编码至多十个新抗原疫苗靶点(SNV 和短插入/缺失)的合成 DNA 片段,通过非免疫原性的甘氨酸/丝氨酸连接子(30 bp)连接,被克隆到起始载体 8 中。该起始载体 8 包含 SEC 结构域 10 (SEC,MRVMAPRTLILLLSGALALTETWAGS)和 MITD 结构域序列 10 (MITD,IVGIVAGLAVLAVVVIGAVVATVMCRRKSSGGKGGSYSQAASSDSAQGSDVSLTA),用于与突变序列串联体融合,以优化其向 HLA I 类和 II 类途径的转运。起始载体还包含 FI 元件 9 (FI,LVLHARNASCPFPVLGTPSLPRPRVPGMLPPPPAPLTTSASSRHL)和骨架序列元件,以提高 RNA 稳定性和翻译效率。 DNA 经 PCR 线性化后,通过分光光度法定量,并用 Sanger 测序法鉴定,然后在洁净室环境下,在 ATP、CTP、UTP、GTP 和β-S-ARCA(D1)帽类似物的存在下,按照先前描述的方法,用 T7 RNA 聚合酶进行体外转录。RNA 采用磁珠纯化,并通过凝胶电泳和微流控毛细管电泳评估其完整性。进一步的分析包括测定浓度、pH 值、渗透压、效价和内毒素水平。
带净阳离子电荷的脂质体用于与 RNA 复合,形成 RNA-LPX 复合物。阳离子脂质体采用基于乙醇注射技术的专有方案 38 制备,原料为阳离子合成脂质(R) -N , N , N- 三甲基-2-3-二油酰氧基-1-丙胺氯化物(R-DOTMA)和磷脂 1,2-二油酰-sn- 甘油 -3-磷酸乙醇胺(DOPE)(默克公司)。脂质体的释放分析包括外观、脂质浓度、RNase 活性、粒径和多分散指数、渗透压、pH 值、亚可见颗粒、热原检测和无菌性检测。 RNA-LPX 药物产品在符合良好生产规范(GMP)的生产设施中制备。首先将各 RNA 与 14.6% (w/v)氯化钠水溶液(Hospira)孵育,使 RNA 溶液中的氯化钠浓度达到预期值,然后再与脂质体混合。随后,采用专有工艺自动混合预处理的 RNA 和脂质体,从而精确控制两种组分的混合比例。通过添加含有定制缓冲液的冷冻保护剂,将 RNA-LPX 进一步稀释至最终药物产品浓度。将原料药灌装至 I 型玻璃小瓶(Ompi)。检查无可见颗粒后,将药物产品冷冻,并以浓缩分散液的形式运送至医院进行稀释。RNA-LPX 的释放分析包括外观、粒径和多分散性、亚可见颗粒、RNA 完整性、RNA 含量、pH 值、渗透压、内毒素检测和无菌性检测。 给病人服用时,将解冻后的药品用 0.9% 氯化钠水溶液稀释。
用于免疫原性检测的血液采样
为了评估疫苗诱导的免疫反应,分别在基线、接种第四剂、第六剂和第八剂疫苗前以及第八剂疫苗接种后 7-14 天采集血样。治疗结束后,在末次接种疫苗后 49-63 天采集血样。对于积极参与长期随访的患者,在主要研究阶段结束后的第一年每 3 个月采集一次血样,在第二年和第三年每 6 个月采集一次血样。未积极参与长期随访的患者可通过一项研究项目,在日常就诊期间捐献血液用于研究。外周血单核细胞(PBMC)通过 Ficoll-Hypaque(Amersham Biosciences)密度梯度离心法从外周血或白细胞分离样本中分离。
重叠肽
在免疫原性评估中,使用了覆盖新抗原序列且具有 11 个氨基酸重叠的合成 15 肽(称为 OLP 池,纯度>70%)或 8-11 肽(纯度>90%)。所有合成肽均购自 JPT Peptide Technologies GmbH 公司,并用 10% DMSO 溶解至最终浓度为 3 mM。
外周血单核细胞的 IVS
使用微珠(Miltenyi Biotec)从冷冻保存的 PBMC 中分离 CD4 + 和 CD8 + T 细胞。使用编码患者特异性新抗原靶点的 15mer OLP 池构建个体化的 IVS 培养体系,每次扩增最多使用五个靶点。为此,将纯化的 CD4 + T 细胞在快速树突状细胞存在下进行扩增,效应细胞与靶细胞的比例( E : T )为 10:1,并加入相应的肽段。对于 CD8 + T 细胞的扩增,将纯化的 CD8 + T 细胞与去除 CD4+ T 细胞的 PBMC(E:T 比例为 1:10 或 1:20)在 IL-4 和 GM-CSF(各 1.000 U ml -1 )以及相应的肽段存在下进行共培养。在开始体外刺激(IVS)后一天,加入含有 10 U ml⁻¹ IL-2(Proleukin S,诺华公司)和 5 ng ml⁻¹ IL-15(Peprotech 公司)的新鲜培养基。CD8 IVS 培养物还额外添加了 IL-4 和 GM-CSF(各 1000 U ml⁻¹ )。IVS 培养 7 天后,补充 IL-2(10 U ml⁻¹ )。刺激 11 天后,通过流式细胞术分析细胞,并用于 ELISpot 检测。由于样本量有限,仅有 8 例患者在进行 ELISpot 检测前进行了外周血单核细胞(PBMC)的体外刺激。
IFNγ ELISpot
预先包被有 IFNγ 特异性抗体(Mabtech)的多孔滤板(Merck Millipore)用磷酸盐缓冲液(PBS)洗涤,并用含 2% 人血清白蛋白(CSL-Behring)的 X-VIVO 15(Lonza)封闭 1-5 小时。随后,每孔加入 0.5 × 10⁵ 至 3.3 × 10⁵ 个效应细胞,分别用肽(体外实验)或负载肽的自体树突状细胞(体外实验)刺激 16-20 小时。为了分析体外 T 细胞反应,将冷冻保存的 PBMC 在 37°C 静置 2-5 小时后进行 ELISpot 检测。或者,使用 CD4 或 CD8 耗竭的 PBMC 作为 CD8 或 CD4 效应细胞。所有检测均重复两次或三次,并包含阳性对照(抗 CD3 抗体(Mabtech;1:1000))、 葡萄球菌肠毒素 B(Sigma-Aldrich)或 PHA-L(Thermo Fisher)。结合的 IFNγ的显色方法有两种:一种是使用直接与碱性磷酸酶偶联的二抗(ELISpotPro 试剂盒,Mabtech;用于体外实验);另一种是使用生物素标记的抗 IFNγ抗体(Mabtech,1:1000),随后与 ExtrAvidin-碱性磷酸酶(Sigma-Aldrich)孵育(用于体外实验)。接下来,将微孔板与 BCIP/NBT(5-溴-4-氯-3′-吲哚磷酸和硝基蓝四唑)底物(ELISpotPro 试剂盒,Mabtech(用于体外实验)或 Sigma-Aldrich(用于体外实验))孵育。使用 AID Classic Robot ELISpot 阅读器或 CTL ImmunoSpot 系列 S6CORE 分析仪(ImmunoCapture 图像采集软件 v.6.6)扫描板,并使用 AID ELISpot 7.0 软件(AID Autoimmun Diagnostika)或 ImmunoSpot Professional 软件 v.5.4 进行分析。斑点计数显示为每个重复或三次重复的平均值。 在离体实验中,使用基于两种统计检验(无分布重采样)的自研 ELISpot 数据分析工具(EDA),根据先前文献 40,41 ,将每个样本的肽刺激斑点计数与仅用培养基孵育的同一样本的效应细胞(作为阴性对照)进行比较。当 P 值小于 0.05 且斑点计数至少为 7 时,则将该肽(新抗原)的应答定义为阳性应答。下一步,比较接种疫苗前后样本的 ELISpot 数据分析工具应答结果。仅在接种疫苗后样本中应答结果为阳性的新抗原被归类为诱导新生应答。两种新抗原在接种疫苗前样本中诱导了斑点计数的显著增加。这些斑点计数在接种疫苗后样本中增加了 11 倍(扩展数据图 3 ,绿色方框突出显示了增强的应答)。这些反应被归类为“增强”。在 IVS 后,使用内部统计分析工具,将肽刺激的斑点计数与对照肽负载靶细胞的接种前后样本进行比较。T 细胞疫苗反应定义为接种疫苗后斑点计数至少增加两倍。
肽-MHC 多聚体染色
利用携带来自免疫原性新抗原的 9 至 11 个氨基酸肽段的荧光染料偶联肽/MHC (pMHC)多聚体(ImmunAware 公司)鉴定突变特异性 CD8 + T 细胞。潜在的最小 I 类表位通过 NetMHCpan 预测器(DTU Health Tech 公司)进行鉴定。或者,使用 easYmer 技术(easYmer 试剂盒,ImmuneAware Aps 公司)生成肽-MHC 复合物,并根据制造商的说明,通过基于微珠的流式细胞术检测验证复合物的形成。对于四聚体化,添加了链霉亲和素(SA)-荧光染料偶联物:SA-BV421、SA-BV711、SA-PE、SA-PE-Cy7 或 SA-APC(均来自 BD Biosciences 公司)。首先对细胞进行多聚体染色,然后进行细胞表面标志物染色,如下(括号内为抗体克隆和稀释度):CD28(CD28.8,1:25)、CD197(150503,1:50)、CD45RA(HI100,1:100)、CD3(SK7,1:100)、CD16(3G8,1:100)、CD14(MφP9,1:50)、CD27(L128,1:50)、CD279(EH12,1:25)、CD127(HIL-7R-M21,1:50)和 CD8(RPA-T8,1:50),均购自 BD Biosciences;CD19(HIB19)和 CD4(OKT4)购自 Biolegend。 TCF-1(C63D9,Cell Signaling)在细胞透化后进行胞内染色。我们还使用 4′,6-二脒基-2-苯基吲哚(DAPI;BD)或可固定活细胞染料 eFluor 780 或 eFluor 506(eBioscience)进行活死细胞染色。在 CD3 + (或 CD8 + )、CD4 − CD14−CD16 − CD19 − 或 CD3 + (或 CD8 + )CD4 − 细胞中鉴定出单细胞、活细胞 、 多聚体阳性事件(扩展数据图 7a )。为了检测 IVS 后抗原特异性 T 细胞,我们对单细胞、活细胞、CD3 + 、CD8 + 多聚体阳性淋巴细胞进行设门。
细胞内细胞因子染色
将外周血单核细胞 (PBMC) 与肽在 37 °C 下孵育约 16 小时,同时加入布雷菲德菌素 A 和莫能菌素。使用可固定活细胞染料 eFluor 780(eBioscience,1:1667)对细胞进行活细胞染色,并检测细胞表面标志物 CD8(RPA-T8,1:33)、CD16(3G8,1:100)、CD14(MφP9,1:50)(均购自 BD Biosciences)、CD19(HIB19,1:50)或 CD4(OKT4,1:25)(均购自 Biolegend)。细胞透化后,使用抗 IFNγ(B27,BD Biosciences,1:100)和 TNF(Mab11,BD,1:167)抗体进行细胞内细胞因子染色。在预先设门的单个、活的和 CD14 − CD16 − CD19 − (并非所有实验都使用)群体中,CD8 + 和 CD4 + 细胞中鉴定出了 IFNγ + 和 TNF + 事件(扩展数据图 7b )。
使用 LSR Fortessa SORP 或 FACSCanto II 细胞分析仪(BD Biosciences)进行采集,并通过 FlowJo 软件(Tree Star)进行分析。
肿瘤组织 TCR 谱分析
按照“下一代测序”中所述的方法,从 FFPE 肿瘤组织中提取 DNA。由 iRepertoire 公司使用其专有流程进行测序和生物信息学分析,以获取肿瘤 TCR 克隆型。
血液样本 TCR 测序和生物信息学分析
使用 Qiagen 公司的 RNeasy Mini Spin Columns 试剂盒,从经磁激活细胞分选(MACS)分选的 3–5 × 10⁵ 个 CD8⁺ 或 CD4⁺ T 细胞中提取总 RNA。总 RNA 用 30 μl 水洗脱,所得 RNA 用于批量 TCR 测序。根据制造商手册,使用 Clontech Laboratories(现为 Takara Bio)的 SMARTer Human TCR a/b Profiling 试剂盒构建批量 TCR 谱库。使用 Illumina MiSeq 测序仪和 600 循环 MiSeq Reagent Kit v.3 试剂盒,在 Illumina MiSeq 测序仪上进行双端测序,读长为 2×300 bp。使用 Bcl2Fastq 软件(Illumina)对数据进行解复用。使用开源软件 Trimmomatic v.0.36 对 Fasta 序列进行质量修剪,然后使用开源软件 MiXCR 分析以注释 TCR 序列。将氨基酸水平上互补决定区 3 (CDR3) 相同的 TCR 克隆型合并,并计算其计数和频率值。使用内部脚本计算 TCR 库统计数据。以 TRB CDR3 链氨基酸序列为分子标识符,追踪新抗原特异性 T 细胞克隆在肿瘤组织和分析数据中的变化。
单细胞测序和读取处理
用 MACS 分选的 CD8 + 或 CD4 + 细胞用含 0.04% BSA 的 1 ml Dulbecco’s PBS (DPBS)洗涤一次,使用宽口径离心头以 300 g 离心 4 分钟,然后重悬至 5.3 × 10 5 个细胞/ml。取 2 万个细胞(37.8 µl)转移至新的离心管中,并按照制造商说明书(Chromium Next GEM 单细胞 V(J)J 试剂盒 v.1.1 用户指南,10x Genomics,CG000207 Rev. D)制备 TCR VDJ 文库和基因表达分析文库。使用 Illumina MiSeq 测序仪和 MiSeq 试剂盒(v.3,600 个循环)进行 TCR 文库测序,采用双端测序,2×150 bp 读长。使用 NextSeq 1000/2000 P2 试剂,在 Illumina NovaSeq 测序仪上对 5′基因表达文库进行测序,循环数为 100,版本为 v.3,采用双端测序,读长为 26×91 bp。使用 Cell Ranger 软件(10x Genomics)处理原始测序数据,生成克隆型数据和基因表达的原始计数矩阵。对于 VDJ 数据,Cell Ranger 的输出文件被重新格式化为我们内部数据分析流程兼容的格式,并进行过滤,仅保留包含 TRA 和 TRB 链的克隆型。含有两条 TRB 链的克隆型被丢弃。
RevImMo 分析
利用疫苗接种前后 CD8 + (和 CD4 + )T 细胞的 TCR 谱分析数据,计算每种 TRB CDR3 克隆型在治疗后的克隆富集度。对于治疗前未检测到的新生克隆,通过赋予其与相应 TCR 库中检测到的最低频率相对应的任意频率值来计算治疗诱导的富集度。在两个时间点均存在的克隆被称为预存克隆。利用治疗后 CD8 + (或 CD4 + )T 细胞的单细胞 TCR 谱分析数据,获取每种 TRB 克隆型的配对 TRA/TRB 信息。仅对具有配对信息的 TRB 克隆型进行富集度排序,并用于克隆选择(图 3a )。选择富集度最高的预存克隆和新生克隆各 10 至 15 个进行进一步验证。由 Twist Bioscience 公司将筛选出的候选 TCR 的 V(D)J 基因合成后克隆到 pST1-TRAC/TRBC1/TRBC2 载体骨架中。采用 DNA 模板制备 PCR 扩增克隆的 TRA 和 TRB 链的线性化模板,然后用于体外转录 RNA 以验证其特异性。使用优化的 Jurkat-NFAT 报告基因检测方法评估筛选出的候选 TCR 的疫苗新抗原特异性和功能。基因工程改造的 Jurkat T 细胞系表达荧光素酶报告基因,该报告基因由 NFAT 反应元件(NFAT-RE)驱动,并由 TCR 特异性信号级联诱导。NFAT-TCR/CD3 效应细胞购自 Promega 公司,为冷冻保存细胞。细胞系的重新鉴定通过 ATCC 和 Eurofins 公司的短串联重复序列(STR)分析完成。 所有使用的细胞系均经检测未发现支原体污染。通过 CRISPR-Cas9 介导的内源性 TCR 敲除和基于转座子的 CD8 共受体(α 链和 β 链)稳定插入,对所用报告基因细胞进行了优化。将编码 TRA 和 TRB 的 IVT-RNA 电穿孔导入细胞,孵育 20 小时后,将 2 × 10⁴ 个 Jurkat 细胞与 K562 细胞或自体 CD14⁺ 单核细胞以 5:1 的比例共培养于 384 孔板中,每孔加入 25 μl 培养基(RPMI1640 + 10% 非热灭活胎牛血清)。共培养前,K562 细胞已转染编码患者 HLA 等位基因的 IVT-RNA,并加载了新抗原靶肽库。自体 CD14 细胞也加载了新抗原靶肽库。所有检测均在所有患者特异性 HLA 等位基因上测试了患者特异性新抗原靶点(扩展数据图 4 )。6 小时后,向每个孔中加入等体积(25 μl)的荧光素(Bio-Glo,Promega),并使用酶标仪测量荧光素酶活性。不同孔中测得的发光信号对应于 Jurkat 细胞中 TCR 介导的活化水平。对于每个 TCR,计算其发光强度相对于仅转染 TCR 的效应细胞对照组的倍数变化,并使用两倍变化作为阈值来确定特异性 TCR。在部分病例中,对检测结果为阴性的 TCR 进行后续的多聚体染色实验评估。使用该方法确定 P1 中的 CD8 TCR 18 为阳性。
单细胞基因表达分析
使用 Seurat 软件 v.5.1.0 42 分析计数矩阵。利用细胞条形码将细胞与相应的 VDJ 数据关联,仅保留具有关联 VDJ 克隆型的细胞。为去除低质量细胞和双细胞,将表达两种不同 TRB 链、独特基因数量超过 6,000 个但少于 100 个以及线粒体独特分子标识符百分比超过 10% 的细胞排除在分析之外。然后使用 Seurat 软件包 43 的 SCTransform 函数,采用默认参数,对过滤后的数据进行归一化和缩放,并对线粒体计数百分比进行回归。每个样本的数据过滤和归一化均单独进行。然后使用 Harmony 1.2.1 的 RunHarmony 函数整合来自不同时间点和患者的归一化数据。进行主成分分析后,通过检查“肘部图”选择前 14 个主成分。使用 FindNeighbors 和 FindClusters 函数进行聚类分析,分辨率为 0.5。基于使用 Seurat FindAllMarkers 函数鉴定的差异表达基因,确定聚类身份。基于 TRB CDR3 序列,鉴定出新抗原特异性细胞,并选择这些细胞进行后续分析。如上所述,使用前 10 个主成分和 0.3 的分辨率对这些细胞进行重新分析。差异基因表达分析后,合并具有相似表型的聚类。
对多聚体阳性细胞进行单细胞 TCR 测序
将 2.16 pmol 肽负载的 HLA 单体(Immudex)与 0.48 µl ULoad dCODE 葡聚糖(Immudex,偶联特定 DNA 条形码)混合,制备三种肽-HLA 葡聚糖试剂。肽-HLA 单体和葡聚糖在 4℃避光孵育 30 分钟。使用空白葡聚糖作为对照。对于多聚体染色,将 0.8 µl 100 µM 生物素与组装好的肽-HLA 葡聚糖和对照葡聚糖混合,用于 PBMC 的混合染色。PBMC 用 MACS 缓冲液重悬并洗涤。将 10 × 10⁶个活的 PBMC 重悬于 PBS/2%人血清白蛋白(HSA)溶液中,加入 5 µl Human TruStain FcX Block(Biolegend),总体积为 50 µl,然后加入 16.6 µl 葡聚糖混合物进行染色。细胞在 4℃避光染色 10 分钟。加入 50 µl 抗体表面染色混合物(BV480 标记的小鼠抗人 CD8 抗体、BB515 标记的小鼠抗人 CD4 抗体、APC-eFlour780 标记的小鼠抗人 CD16/CD14/CD19 抗体以及 eFlour780 活细胞固定染料)。细胞在 4℃下与抗体混合物孵育 30 分钟。然后用 1 ml PBS/2% HSA 洗涤细胞 3 次,最后重悬于 500 µl PBS/2% HSA 溶液中。首先通过荧光激活细胞分选 (FACS) 对转运通道 (eFlour780) 阴性的 CD8 阳性淋巴细胞群进行设门,从而分选出多聚体阳性细胞。葡聚糖骨架经藻红蛋白 (PE) 标记,因此多聚体特异性 CD8 T 细胞被分离为 PE 阳性细胞群。分选出的多聚体阳性 T 细胞经离心后,重悬于 50 µl PBS/0.04% BSA 溶液中。测定细胞浓度后,将细胞稀释至最终浓度为 5.3 × 10⁵ 个细胞/ml。单细胞 RNA 测序按上述方法进行。 VDJ 和特征条形码文库的制备按照制造商说明书进行(Chromium Next GEM 单细胞 5′ 试剂盒 v.2 [双索引],10x Genomics,CG000511 Rev. B)。VDJ 文库的数据处理如上所述。特征条形码文库使用 Cell Ranger 软件进行处理,以将多聚体条形码分配给测序细胞。基于细胞条形码,将 VDJ 和特征条形码数据合并后,使用内部脚本确定各个 TCR 克隆的抗原特异性。
免疫组织化学染色
为了确定 CD3、CD8 和 MHC-I 的表达,对 FFPE 肿瘤组织 3 至 4 µm 厚的切片进行了分析。
染色在 Ventana Discovery Ultra 平台上进行。抗原修复采用 Ultra CC1 试剂盒,时间为 64 分钟。之后,将玻片与抗人 CD3 抗体(2GV6;Roche Diagnostics,即用型稀释液)、抗人 CD8 抗体(SP57;Roche Diagnostics,1:100)和 MHC-I 抗体(EPR1394Y;Abcam,1:500)在 37°C 下孵育 60 分钟,随后与二抗 OmniMap 抗兔 HRP(Roche Diagnostics,即用型稀释液)在 37°C 下孵育 32 分钟。显色采用 ChomoMap DAB 试剂盒(Roche Diagnostics),随后进行复染(苏木精 II,蓝化试剂;Roche Diagnostics)。玻片经扫描(Axio.Scan;Zeiss)后进行人工分析。
差异基因表达分析、GSEA 和变异等位基因频率计算
使用生物信息学软件流程分析了治疗后样本中 P12 和 P13 的全外显子组测序和 RNA-seq 数据。使用 R (v.4.0.2) 和 tximport (v.1.18) 导入由 Sailfish 生成的 reads 计数,并按基因汇总转录本 reads 计数,对 RNA-seq 数据进行差异表达分析。根据软件包 vignette 中的说明,将汇总的计数导入 DESeq2 (v.1.30) 进行差异表达检验,仅考虑在所有样本中至少有 10 个 reads 覆盖的基因。差异表达检验结果以 0.01 的显著性阈值导出(使用 Benjamini-Hochberg 方法 45 进行错误发现率校正后),并计算收缩后的 log2 倍数变化值。使用 fgsea 1.20.0 46 R 包对 MSigDB v.7.5.1 47 KEGG 经典通路进行 GSEA 分析。根据 Wald 统计值对治疗前后肿瘤差异表达基因进行排序,并将其作为 fgsa 函数的输入,执行 1,000 次置换。使用 pysam (v.0.15.4) 48 , 49 , 50 从比对的肿瘤 BAM 文件中确定治疗前肿瘤中体细胞 SNV 的变异等位基因频率。
Kaplan-Meier 生存分析用于评估无病生存概率
无病生存期是本报告中一项非方案规定的分析。无病生存期定义为从首次接种疫苗到癌症复发或任何原因死亡的时间。初始(新)辅助治疗后的影像学检查仅在怀疑疾病进展时进行,这可能高估了无病生存期。最后一次就诊/联系日期作为截尾日期。该分析使用 SAS 9.4 版软件进行。
材料供应情况
本文材料由作者根据与 BioNTech 签订的材料转移协议提供。本作品采用知识共享署名 4.0 国际许可协议(CC BY 4.0)进行许可,允许在任何媒介上无限制地使用、分发和复制,前提是必须正确引用原始作品。如需查看此许可协议的副本,请访问 http://creativecommons.org/licenses/by/4.0/ 。本许可协议不适用于文章中包含的、注明出处为第三方的图表/照片/艺术作品或其他内容;使用此类材料前,请务必获得版权所有者的授权。
报告摘要
有关研究设计的更多信息,请参阅本文链接的《 自然投资组合报告摘要》 。
数据可用性
与本研究相关的所有数据均已包含在论文或补充材料中。
Hits: 22